


Ch. 17 - Pharmacology of Pain
Jenny Koehl, PharmD, BCPS | Massachusetts General Hospital
Herein we discuss the pharmacology of commonly employed analgesic agents in the emergency department. This information will assist providers in selecting therapies with different pharmacologic mechanisms to target multiple pain pathways, reduce opioid usage, and avoid periods of inadequate pain relief. Within this strategy, around the clock dosing of medications is important to prevent the undertreatment of pain as well as lower the dose of medications overall.
ALTERNATIVES TO OPIOIDS (ALTO)
Acetaminophen
Acetaminophen (APAP) has antipyretic and analgesic effects but lacks peripheral anti-inflammatory properties. The antipyretic and a portion of the analgesic effects result from selective inhibition of a recently discovered COX-1 splice variant enzyme, COX-3, present in the brain and spinal cord.1 COX-1 is constitutively expressed and is responsible for maintaining gastrointestinal (GI) mucosa, regulating renal blood flow, and platelet aggregation.2 APAP easily crosses the blood brain barrier and is distributed throughout the central nervous system (CNS) allowing it to reach concentrations in the brain able to inhibit COX-3 enzymatic activity. APAP does not bind directly to the active site on the enzyme but reduces the active form and inhibits its catalytic activity.3
In addition to centrally-mediated COX inhibition, analgesic and antipyretic properties of APAP also stem from modulation of the endogenous cannabinoid system via the vanilloid and cannabinoid signaling.4 After de-acetylation to p-aminophenol, APAP undergoes conjugation with arachidonic acid to form N-arachidinoyl-phenolamine (AM404) in the CNS.5 The structures of AM404 and endogenous cannabinoid neurotransmitters are similar which allows the APAP metabolite to act as a weak agonist of cannabinoid receptors leading to increased levels of endogenous cannabinoids.6-8 AM404 is also a potent activator of the vanilloid subtype 1 receptors whereby causing vasodilation.9
Acetaminophen is often administered with ibuprofen for acute pain presentations in the emergency setting where the combination of acetaminophen (1,000 mg) plus ibuprofen (400 mg) simultaneously was found to be superior to using either agent alone.47 A 2017 study found the combination of acetaminophen/ibuprofen was as efficacious as the opioid-containing combinations oxycodone, hydrocodone, codeine plus acetaminophen when used for musculoskeletal pain in an emergency setting reinforcing its use over opioids in mild and moderate pain settings.91
Acetaminophen is listed as a Category B FDA rating for the first through third trimester of pregnancy secondary to a favorable safety profile (Black and Hill 2003; de Fays et al. 2015). In mild to moderate pain, acetaminophen is considered the safest first line analgesic for pregnancy and lactation.94
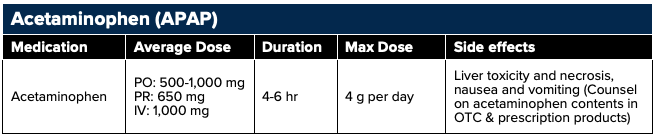
Dosing: Daily doses of APAP should not exceed 4,000 mg or 3,000 mg in patients with a history of liver disease or the elderly. APAP can be given orally (PO), per rectum (PR), or intravenously (IV), although the IV preparation is expensive and lacks data to suggest superior clinical efficacy.10
Non-steroidal anti-inflammatory drugs
Non-steroidal anti-inflammatory drugs (NSAIDs), unlike APAP, are competitive inhibitors of COX-1 and COX-2 which mediate the conversion of arachidonic acid to inflammatory prostaglandins. NSAIDs reduce the levels of prostaglandins in the central and peripheral nervous system, relieving pain, swelling and redness. NSAIDs also inhibit prostaglandin-mediated stimulation of the hypothalamus which results in a lowering of body temperature.2
Traditional NSAIDs block prostaglandin synthesis via non-selective inhibition of COX-1 and COX-2 isozymes. These agents include aspirin, ibuprofen, naproxen, and ketorolac. Within this non-selective class, aspirin is unique in that it irreversibly inhibits the COX enzymes, whereas the rest of the class competitively inhibits the active site. This results in aspirin's platelet aggregation inhibition lasting the lifespan of the cell. As COX-1 is responsible for maintaining gastrointestinal (GI) mucosa, regulating renal blood flow, and platelet aggregation, side effects of the non-selective NSAIDs include nausea and vomiting, gastric ulceration, bleeding, and kidney injury.11 There are a few agents that are non-selective but have a higher affinity for the COX-2 enzyme and these include indomethacin, meloxicam, and diclofenac. Second generation NSAIDs are selective COX-2 inhibitors, with the COX-2 isoenzyme only expressed during times of inflammation. This selectivity results in an improved gastric safety profile; however, COX-2 selectivity has shown to increase risk of myocardial infarction, stroke, and heart failure resulting from induction of a pro-thrombotic state.12 Celecoxib is the only COX-2 selective NSAID currently available on the market as other agents have been removed due to their increased cardiovascular (CV) risks.
Summary: COX-3 is associated with centrally mediated analgesic and antipyretic properties, but low anti-inflammatory activity. COX-1, the precursor to COX-3 has similar properties but may be targeted centrally and peripherally. COX-2 is involved with the inflammatory process and NSAIDs that inhibit COX-2 are best for pain resulting from inflammatory states.
When selecting an NSAID one must weigh the GI, renal, and CV risks. If the patient is at higher risk of a GI bleed consider adding a proton pump inhibitor to reduce risk of GI bleed.
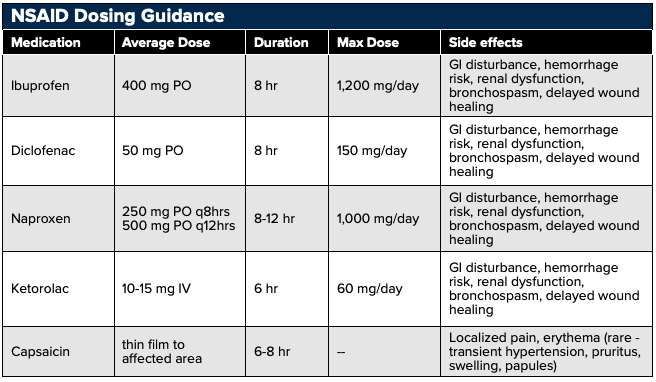
All NSAIDs have an analgesic ceiling dose, which is lower than the anti-inflammatory maximal dose. When the ceiling dose is met, additional increases in dose provide no further analgesic benefit. This ceiling dose has been reported as 1,0000 mg for aspirin and acetaminophen, 400 mg for ibuprofen, and 10 mg for IV ketorolac.13-15 Yet higher doses may be needed to achieve additional anti-inflammatory benefits. With this in mind, lower dosages should be selected for noninflammatory pain, and higher ranges are reserved for those situations in which inflammation and swelling are significant contributors to pain.
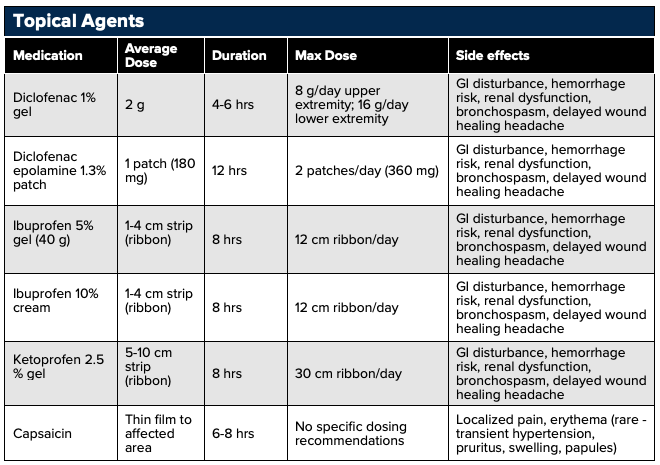
Topical NSAIDs may also be considered. As opposed to PO and IV analgesics which require systemic distribution, topical/transdermal analgesics use localized analgesic distribution to limit total-body quantities delivered.76,109,113 Topical NSAIDs can preferably accumulate in targeted areas such as cartilage and meniscus with concentrations 4-7 times greater than plasma concentrations, and in tendons with concentrations one hundred times greater than plasma concentrations.76 Topical analgesics are optimal in patients with renal disease or elderly patients susceptible to elevated analgesic plasma concentrations and in patients with multiple comorbidities such as peptic ulcer disease and cardiovascular disease in which PO NSAIDs are relatively contraindicated.85
Examples of topical analgesics include diclofenac, ketoprofen, and ibuprofen. Pain pathologies that benefit from topical analgesics include acute sprain, strain, overuse injuries, contusions, tendinopathies, bursitis, exacerbations of osteoarthritis.107 Adverse effects include milder GI disturbance, hemorrhage risk, renal dysfunction, bronchospasm, delayed wound healing headache.110
Sub-dissociative Ketamine
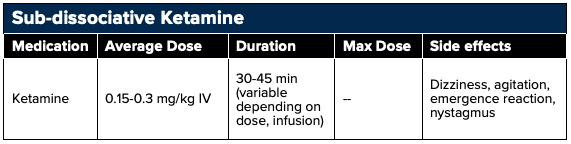
Ketamine is an analgesic adjunct often used for pain management in the setting of intractable pain, neuropathic pain, or opioid-tolerant and opioid-induced hyperalgesic states. Once in the blood stream ketamine redistributes to the CNS in about 30-45 seconds resulting in a very rapid onset of action. When using ketamine for pain a sub-dissociative dose of 0.1–0.3 mg/kg IV as a single dose given over 10-15 minutes, 0.15 mg/kg/hr as a continuous infusion, or 0.7–1 mg/kg intranasal should be employed.16 Ketamine is highly water soluble with high lipophilicity and should be dosed based on ideal body weight.
At these lower doses, ketamine acts as a noncompetitive N-methyl-D-aspartate (NMDA) receptor antagonist in the brain and spinal cord24 as well as a partial mu receptor agonist. This results in pain relief as the NMDA receptor is involved in the amplification of pain signals, the development of central sensitization, and opioid tolerance.23 Additionally, ketamine has been shown to have anti-hyperalgesic effects that may reduce or reverse opioid tolerance.25,26
Ketamine's adverse effects are dose-dependent. At sub-dissociative doses ketamine may result in psychoperceptual side effects, a feeling of unreality, sedation, nausea, vomiting, dizziness, and nystagmus (seen shortly after onset). These adverse effects have been shown to be decreased when administered as an infusion over 10-15 minutes rather than a bolus dose.17 Selecting doses at the lower end of the dosing range will minimize adverse effects particularly in the geriatric population (Motov, 2018). At higher doses ketamine also acts on dopamine2 receptors, monoaminergic receptors, monoamine transporters, and opioid receptors, which results in psychotomimetic affects including hallucinations, agitation, and dysphoria.22
Sodium-Channel Blockers
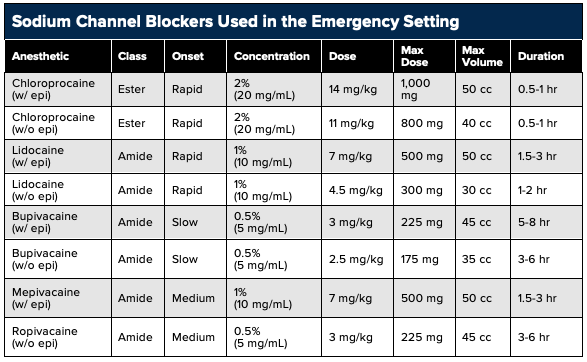
Sodium channel blockers function as analgesics through the noncompetitive inhibition of sodium channels and, consequently, of nerve signal propagation.19,20 Sodium channel blockers consist of two classes: esters (chloroprocaine, procaine, tetracaine) and amides (lidocaine, mepivacaine, ropivacaine, bupivacaine). Whereas ester bonds are more rapidly hydrolyzed, amide bonds are more resistant to clearance and thus offer a longer duration of action. Historically lidocaine has been utilized for the treatment of chronic neuropathic pain, but more recently lidocaine has been studied for renal colic, headache, and in the perioperative setting.21 The expanse of utility stems from lidocaine’s analgesic, anti-hyperalgesic, and anti-inflammatory properties. Lidocaine is a sodium channel blocker that slows the flow of sodium ions across the cell membrane, lessening the influx of calcium ions into the nerve terminals and inhibiting the release of the excitatory neurotransmitter “glutamate.” This reduces spontaneous nerve firing, causing a decrease in the sensation of pain.41
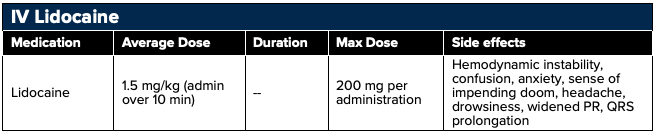
Intravenous lidocaine can be given as a bolus of 1-2 mg/kg (maximum 200 mg) or as a continuous infusion of 0.5-3 mg/kg/hr based on ideal body weight.28 Administering the bolus dose prior to initiating the continuous infusion may result in faster time to therapeutic effect.
Lidocaine is a Vaughn Williams Class IB antiarrhythmic and there is the potential of developing cardiac arrhythmias. Although the risk is low it does increase with higher dosages and accumulation in the setting of liver dysfunction. More common adverse effects include dizziness and paresthesias.27 Serum levels of lidocaine can be obtained; however, this is a send-out laboratory test for many hospitals resulting in a lag time of 48 hours or greater. If lidocaine levels are obtained, the desired concentration to avoid unwanted side effects is less than 4 mcg/mL.
Dopamine Receptor (D1-R, D2-R) Antagonist
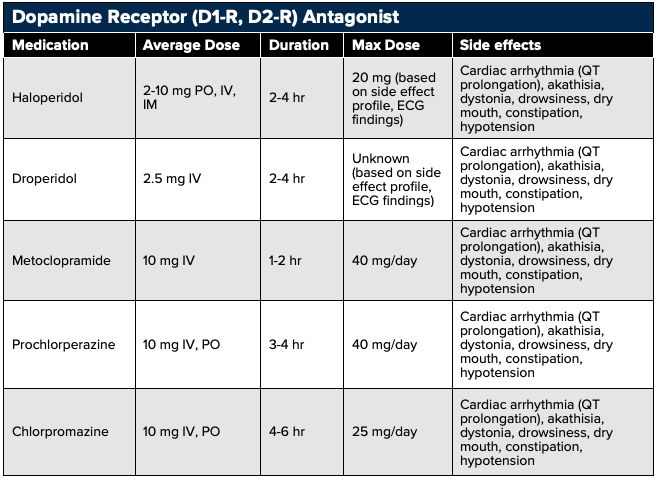
Dopamine receptor antagonists including metoclopramide, prochlorperazine, chlorpromazine, haloperidol, and droperidol provide analgesia through the modulation of the dopamine-centered pain signaling pathways.29
Metoclopramide, prochlorperazine, chlorpromazine, and droperidol has all been shown to effectively treat acute migraines.30-34 Droperidol and haloperidol have also been shown to be efficacious in the treatment of gastroparesis, cannabinoid-induced hyperemesis, and cycling vomiting syndrome.35,36
Side effects of dopamine-receptor antagonists include QT prolongation, extrapyramidal side effects (akathisia, dystonia), anti-muscarinic effects (drowsiness, dry mouth, constipation, hypotension), and neuroleptic malignancy syndrome (hyperpyrexia, muscle rigidity, rhabdomyolysis) (Vinson, 2001; D’Souza, 2018).
Metoclopramide, prochlorperazine, and chlorpromazine can be administered over a 15 to 30 minute infusion to reduce extrapyramidal effect (Bigal, 2002; D’Souza, 2018). 25 mg IV or PO diphenhydramine may also be used with prochlorperazine to offset the associated akathisia.37,38
Dexmedetomidine
The alpha-2-adrenergic receptor agonist dexmedetomidine produces analgesia by dampening the centrally-activated sympathetic (adrenergic) pathway.39 Dexmedetomidine utilization has expanded from it classic role as a sedative among mechanically ventilated patients in intensive care settings, to a promising analgesic adjunct outside the ICU.39 Concomitant DXMT use has been shown to reduce opioid (oxycodone) consumption, decrease opioid side effect profile, and improve patient satisfaction in the postoperative setting.40 Dexmedetomidine can be delivered IV or IN with recommended dosing of 0.5 to 1.0 μg/kg IV (1 to 2 μg/kg intranasal).39 At this time of this review, a high cost and reduced availability in the has limited research and utilization of DXMT in the emergency setting and its use remains an emerging concept inneed of further research.

Adjunct Agents for Neuropathic Pain
Anticonvulsants
Gabapentin and pregabalin are effective treatments for post hepatic neuralgia, phantom limb pain, peripheral neuropathy, and pain caused by nerve compression. Gabapentin and pregabalin have the same binding site on the α2-δ subunit of presynaptic, voltage-dependent calcium channels that are located throughout the peripheral and central nervous systems. Similar to lidocaine, blocking the calcium channels reduces the flow of calcium ions into the nerve terminals and inhibits the release of the excitatory neurotransmitter "glutamate" and nerve cell excitability.42 A common misconception is that these agents alter GABA uptake and degradation however, they are inactive at GABAA and GABAB receptors, and are not converted metabolically into GABA or a GABA antagonist.
Although gabapentin and pregabalin have the same pharmacologic profile, the binding affinity for the α2-δ subunit, and potency, of pregabalin is six times more than that of gabapentin.43 Gabapentin also has variable interindividual bioavailability with saturable oral absorption, meaning that its bioavailability decreases as the dose increases (Neurontin PI). Therefore, smaller doses, given more frequently, may be required to optimize absorption. Pregabalin has a more predictable, linear pharmacokinetic profile that is not saturable (Pregabalin PI).
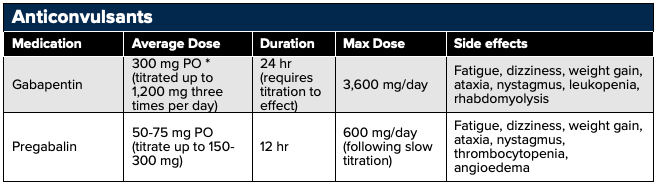
Important: Gabapentin and pregabalin may be initiated in the emergency department but the onset of pain relief is not typically seen immediately. These agents require a slow titration to effect over several weeks. As a result, they should be started at a low dose and titrated based upon clinical effect, renal function, and presence of adverse effects by an outside provider. Additionally, neither gabapentin nor pregabalin should be administered with opioids as both may potentiate the euphoric effects of opioids when taken concomitantly, increasing susceptibility to abuse (100) and a worsening respiratory and CNS depression.
Example Dosing/Titration/Therapeutic switch of Pregabalin
- Dosing in diabetic peripheral neuropathy
- Begin at 50 mg PO three times daily and increase to 100 mg three times daily within 1 week based on efficacy and tolerability.
- Dosing in postherpetic neuralgia
- Begin at 75 mg PO twice a day or 50 mg three times daily and increase to 100mg three times daily within 1 week based on efficacy and tolerability. Dosing may be further escalated over 2–4 weeks to a maximum of 300 mg twice a day, or 200 mg three times daily.
- Converting to Pregabalin: Gabapentin should be discontinued over a minimum of 1 week before starting pregabalin at a dose of 50 mg three times daily. You may also add pregabalin to gabapentin during the titration, but side effects may be additive.
Antidepressants
Antidepressants may be used to treat chronic neuropathic pain typically at lower doses than those given for antidepressant effects. Tricyclic antidepressants (TCAs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) have the most available data.
TCAs with SNRI activity including amitriptyline (tertiary amines) and nortriptyline (secondary amine) as well as the SNRI inhibitor duloxetine are commonly used for neuropathic pain. The exact mechanism of pain relief is not fully understood, but the theory is that the descending inhibitory pathways are potentiated by inhibiting the reuptake of serotonin and norepinephrine in the spinal synapses between first-order and second-order neurons.47,48 Additionally, serotonin’s activation of metabotropic receptors and norepinephrine’s binding to alpha adrenergic receptors can increase the release of endogenous opioids.49
Caution should be taken with the TCAs, especially in the elderly population, due to their strong anticholinergic activity resulting in dizziness, drowsiness, dry mouth, nausea, constipation, and cardiotoxicity, particularly in the elderly population. Additional adverse effects include paresthesias, flushing, palpitations, fatigue, transient neck tightness, chest pressure, and dysrhythmias.94,95
Triptans
Triptans have high agonist activity on 5-HT1b,1d receptors resulting in vasoconstriction of intracranial blood vessels, inhibition of vasoactive neuropeptide release, and blocking transmission of pain signals.50-53 The pharmacokinetic properties of the triptans differ depending on their bioavailability, CNS penetration, route of administration, and metabolic pathways. However, pharmacokinetic differences have shown little impact on clinical efficacy, but may influence the preference of the patient.64,65 Therefore, the American Academy of Neurology supports the role of all the triptans in the abortive management of moderate-to-severe migraines.66 (Silberstein, 2000), and if the patient fails or develops intolerance to one triptan a trial of an alternative agent should be performed.67
Contraindications to the triptans stem from their vasoconstrictive properties including cardiac ischemia that has been reported in patients with and without a CV history.127 Although unclear whether the etiology is a result of a true vascular pathology, the triptans are contraindicated in patients with coronary artery disease, cerebrovascular disease, uncontrolled hypertension, rhythm disturbances, peripheral vascular disease, ischemic bowel disorders, and hemiplegic or basilar migraine (Triptan PIs).
Topical Analgesic Agents
Topical/transdermal preparations are an attractive option to reduce systemic adverse effects of medications while providing pain relief for conditions such as sprains, strains, tendinopathies, as well as several neuropathic conditions like post-herpetic neuralgia, burns, and arthritic flares. In addition to limited system absorption that ranges from 0.2% to 8% of the oral route, topic NSAIDs can accumulate in cartilage and tendons resulting in higher localized concentrations.76 Keep in mind that topical preparations can only penetrate 8–10 mm and are most useful in well-localized neuropathic and inflammatory pain.
Examples of topical analgesic agents include:
- Lidocaine topical: 2-5%; cream, gel, ointment
- Lidocaine patch: 5% prescription patch or 4% OTC patch
- Capsaicin patch (8%) and cream (0.025 to 0.075%)
- Diclofenac: gel, solution, patch
- Ketoprofen
- Methyl salicylate-menthol ointment
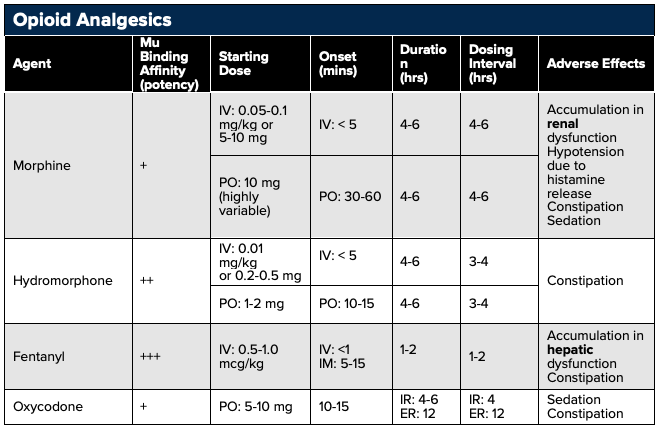
Opioid Analgesia
Opioids may be used in the treatment of traumatic injuries, visceral pain, vaso-occlusive crisis, and cancer related pain. Opioids mimic the body’s own analgesic system with a similar molecular structure as endogenous opioids.
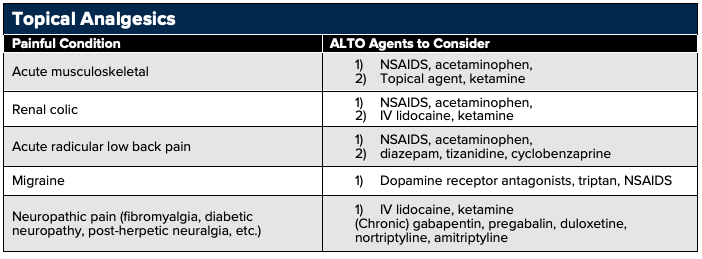
There are 3 classes of opioids, with the naturally occurring category derived from opium. Semi-synthetic opioids have been chemically modified from the natural opiates, and synthetic opioids are entirely chemically manufactured. Knowing the classification of agents may help with selection when faced with patient reported opioid allergies or intolerances.
There are three types of opioid receptors: mu, kappa, and delta with the mu receptor primarily responsible for the analgesic effect. Of note, tramadol also inhibits reuptake of serotonin and norepinephrine in addition to weak mu receptor binding.
Activation of the mu receptor also triggers respiratory depression and physical dependence, which have not been associated with delta and kappa receptor activation.77 Opioids can also activate mast cell degranulation leading to histamine release and subsequent flushing, urticaria, pruritus, and/or hypotension. However, not all opioids produce the same level of histamine release with morphine and codeine having the highest and fentanyl and hydromorphone inducing little to no release.
Morphine
Morphine is considered the gold standard and the baseline for which all other opioids are measured.121 Similar to other opioids, morphine derives its effects by preferentially activating the mu-opioid receptors (MOR).103,111,126
Morphine undergoes significant first-pass metabolism in the liver with 20-25% overall oral bioavailability.104 Compared with more lipid-soluble opioids such as codeine, heroin, and methadone, morphine crosses the blood-brain barrier at a considerably lower rate.95 Morphine is metabolized by the liver through glucuronidation (UDP glucuronosyltransferase, UGT2B7) to active metabolites morphine-6-glucuronide (M6G) and morphine-3-glucuronide (M3G).92 M6G is a more potent analgesic version of morphine.115,117 M3-G has less affinity for opioid receptors but causes neuroexcitatory effects (allodynia, myoclonus, seizures) (Smith 2000). Morphine metabolism is independent of the CYP-mediated metabolism, effectively avoiding these drug-drug metabolism interactions.122 However, similar to the allelic variation seen with CYP2D6 enzymatic activity, variability exists among UGT2B7 enzyme metabolism and should be considered in patients who appear ultrasensitive, or conversely unresponsive, to standard doses of morphine.88,93,98
Following metabolism, morphine metabolites are renally excreted with a small amount excreted as the parent compound.97,105 Morphine metabolites (M3G and M6G) may accumulate in renal failure resulting in significant toxicity.97 Morphine should be used cautiously in patients with renal disease and elderly patients with decreased renal function.97,105,116,118
Fentanyl
Fentanyl is metabolized by the liver through dealkylation (CYP3A4) producing inactive metabolites that are renally excreted.101,119 Drug-drug interactions can occur secondary to competition for CYP3A4 metabolism. Fentanyl should be used cautiously with CYP3A4 competing substrates and inhibitors like statins, amiodarone, haloperidol, macrolides, azole antifungals (fluconazole, ketoconazole), calcium channel blockers, grapefruit juice (bergamottin), methylprednisolone, and protease inhibitors (indinavir, ritonavir).120,122 The metabolism of fentanyl to inactive metabolites allows for safe use in patients during renal failure or dialysis.97
In addition to the common opioid side effects such as respiratory depression, fentanyl toxicity can result in muscle (chest wall) rigidity.87,90 In contrast to other opioids, fentanyl does not result in histamine-induced hypotension making its use safe in hemodynamically compromised patients.114
Hydromorphone
Similar to oxymorphone and morphine sulfate, hydromorphone (HM) is independent of CYP450 metabolism, avoiding these drug-drug interactions.122,125 HM undergoes hepatic glucuronidation producing the primary metabolite, hydromorphone-3-glucuronide (H3G). Similar to morphine metabolite M3G, H3G is neuroexcitatory and excessive buildup can lead to myoclonus and seizure activity.122-124
HM and its metabolites are renally excreted and should be cautiously administered in patients with renal disease or dialysis.97,105,112 HM metabolites are more efficiently dialyzed than morphine metabolites and thus better tolerated among patients on dialysis.97,105
Side effects of HM are dose-dependent and similar to general opioids.112 If pruritus becomes intolerable, administration of oral diphenhydramine, not IV, may be considered as the latter potentiates sedation while offering limited pruritic relief.106
Oxycodone/ Hydrocodone
Oxycodone (OC) and hydrocodone (HC) were both marketed with the advantage of reduced first pass metabolism over oral morphine.99 Both OC and HC are metabolized in the liver (CYP2D6, CYP3A4) to their active metabolites oxymorphone and hydromorphone.122 Metabolic variability exists among these enzymes resulting in ineffective analgesia in the “poor metabolizers” and toxic levels in the “ultra-rapid metabolizers”.99,100 Similar to morphine, the active metabolites of oxycodone and hydrocodone accumulate in renal failure and should be used cautiously in patient with renal disease and elderly patients with decreased renal function.97,105,116,118
Codeine
Codeine is a prodrug with analgesic effects dependent on the metabolic conversion from the prodrug form to codeine-6-glucuronide (C6G) and morphine by the liver CYP450 enzyme CYP2D6 (similar to tramadol). This enzymatic reaction has significant allelic variability.102,122 Both poor and ultra-rapid metabolizers exist among the population with up to 10% of population lacking the enzyme necessary for codeine conversion and 1-7% of the Caucasian population being ultrarapid codeine metabolizers.86,99,102,108 This variation results in uncontrolled variability in analgesic response. Nursing mothers may also produce breast milk containing higher than expected levels of metabolites that can lead to severe adverse events in nursing infants.96
In addition to varying side effect profiles, onset of action and potency differs between opioid analgesics. For example, morphine has low lipid solubility and slow blood-brain barrier penetration resulting in a slow onset of action. On the other hand, fentanyl is highly lipid soluble, resulting in a very rapid onset of action.78 Furthermore, hydromorphone and fentanyl exhibit the highest mu receptor affinity resulting in the greatest analgesic effects.
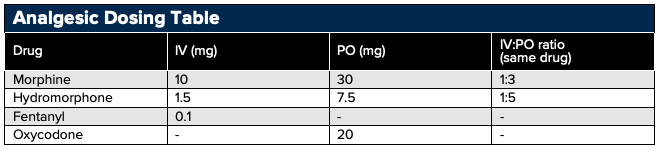
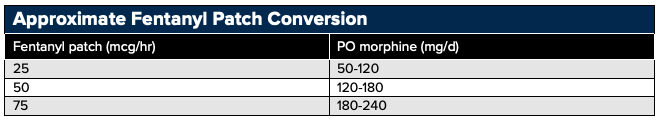
Opioid Conversion Tips
- First calculate the morphine equivalent of the opioid being converted from, and then calculate the dose of the opioid being converted to based on the morphine equivalent
- When converting patients from one opioid to another decrease the dose of the new opiate by 25-50% (after calculations) due to incomplete cross-reactivity between opiates
References
- Chandrasekharan NV, Dai H, Roos KL, et al. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyreticdrugs: cloning, structure, and expression. Proc Natl Acad Sci U S A. 2002;99(21):13926-31.
- Dubois RN, Abramson SB, Crofford L, et al. Cyclooxygenase in biology and disease. FASEB J. 1998;12(12):1063-73.
- Boutaud O, Aronoff DM, Richardson JH, et al. Determinants of the cellular specificity of acetaminophen as an inhibitor of prostaglandin H(2) synthases. Proc Natl Acad Sci U S A. 2002;99:7130–7135.
- Ghanem CI, Perez MJ, Manatuou JE, et al. Acetaminophen from the liver to brain: New insights into drug pharmacological action and toxicity. Pharmacol Res. 2016;109:119-31. doi: 10.1016/j.phrs.2016.02.020.
- Hogestatt ED, Jonsson BA, Ermund A, et al. Conversion of acetaminophen to the bioactive N-acylphenolamine AM404 via fatty acid amide hydrolase-dependent arachidonic acid conjugation in the nervous system. J Biol Chem. 2005;280:31405–31412.
- Beltramo M, Stella N, Calignano A, et al. Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science. 1997;277:1094–1097
- De Petrocellis PL, Bisogno T, Davis JB, et al. Overlap between the ligand recognition properties of the anandamide transporter and the VR1 vanilloid receptor: inhibitors of anandamide uptake with negligible capsaicin-like activity. FEBS Lett. 2000;483:52–56.
- Szallasi A, Di Marzo V. New perspectives on enigmatic vanilloid receptors. Trends Neurosci. 2000;23:491–497.
- Zygmunt PM, Chuang H, Movahed P, Julius D, Hogestatt ED. The anandamide transport inhibitor AM404 activates vanilloid receptors. Eur J Pharmacol. 2000;396:39–42.
- Furyk J, Levas D, Close B, et al. Intravenous versus oral paracetamol for acute pain in adults in the emergency department setting: a prospective, double-blind, double-dummy, randomised controlled trial. Emerg Med J. 2018; 35:179-84.
- Bhala N, Emberson J, Merhi A, et al. Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: meta-analyses of individual participant data from randomised trials. Lancet 2013;382:769-79.
- Solomon SD, McMurray JJ, Pfeff er MA, et al. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. New Engl J Med 2005; 352: 1071–80.
- Troullos ES, Freeman RD, Dionne RA. The scientific basis for analgesic use in dentistry. Anesth Prog. 1986;33:123–138.
- Laska EM, Sunshine ., Marrero I, et al. The correlation between blood levels of ibuprofen and clinical analgesic response. Clin Pharmacol Ther. 1986;40:1–7.
- Motov S, Yasavolian M, Likourezos A, et al. Comparision of intravenous ketorolac at three single-dose regimens for treating acute pain in the emergency department: a randomized controlled trial. Ann Emerg Med. 2017; 2017:177-84.
- Motov S, Rosenbaum S, Vilke GM, Nakajima Y. Is there a role for intravenous subdissociative-dose ketamine administered as an adjunct to opioids or as a single agent for acute pain management in the emergency department? J Emerg Med. 2016; 51:752-57.
- Motov S, Mai M, Pushkar I, et al. A prospective randomized, double-dummy trial comparing IV push low dose ketamine to short infusion of low dose ketamine for treatment of pain in the ED. Am J Emerg Med. 2017;35:1095-1100.
- Motov S, Mann S, Drapkin J, et al. Intravenous subdissociative-dose ketamine versus morphine for acute geriatric pain in the emergency department: a randomized controlled trial. Am J Emerg Med. 2019;37(2):220-227.
- Golzari SE, Soleimanpour H, Mahmoodpoor A, et al. Lidocaine and pain management in the emergency department: a review article. Anesth Pain Med. 2014;4(1):e15444.
- Sinnott CJ, Cogswell IL, Johnson A, et al. On the mechanism by which epinephrine potentiates lidocaine's peripheral nerve block. Anesthesiology. 2003;98(1):181-8.
- Masic D, Liang E, Long C, et al. Intravenous lidocaine for acute pain: a systemic review. Pharmacotherapy. 2018;38(12):1250-1259.
- Bell RF, Kalso EA. Ketamine for pain management. Pain Rep. 2018;3(5):e674.
- Trujillo KA, Akil H. Inhibition of morphine tolerance and dependence by the NMDA receptor antagonist MK-801. Science 1991;251:85–7.
- Milon G, Villevieille T. Ketamine pharmacology: an update (pharmacodynamics and molecular aspects, recent findings). CNS Neurosci Ther. 2013;19(6):370-80.
- Laulin JP, Maurette P, Corcuff JB, et al. The role of ketamine in preventing fentanyl-induced hyperalgesia and subsequent acute morphine tolerance. Anesth Analg 2002;94:1263–69.
- Lilius TO, Jokinen V, Neuvonen MS, et al. Ketamine coadministration attenuates morphine tolerance and leads to increased brain concentrations of both drugs in the rat. Br J Pharmacol 2015;172:2799–813.
- Silva LO, Scherber K, Cabrera D, et al. Safety and efficacy of intravenous lidocaine for pain management in the emergency department: a systematic review. Ann Emerg Med. 2018; 72(2): 135-144.
- Soleimanpour H, Hassanzadeh K, Vaezi H, et al. Effectiveness of intravenous lidocaine versus intravenous colic in the emergency department. BMC Urol. 2012; 12:13. Doi: 10.1186/1471-2490-12-13.
- Wood PB. Role of central dopamine in pain and analgesia. Expert Rev Neurother. 2008;8(5):781-97
- Friedman BW, Esses D, Solorzano C, et al. A randomized controlled trial of prochlorperazine versus metoclopramide for treatment of acute migraine. Ann Emerg Med. 2008;52(4):399-406.
- Friedman BW, Irizarry E, Solorzano C, et al. Randomized study of IV prochlorperazine plus diphenhydramine vs IV hydromorphone for migraine. Neurology. 2017;89(20):2075-82.
- Bigal ME, Bordini CA, Speciali JG. Intravenous chlorpromazine in the emergency department treatment of migraines: a randomized controlled trial. J Emerg Med. 2002;23(2):141-8.
- Weaver CS, Jones JB, Chisholm CD, et al. Droperidol vs prochlorperazine for the treatment of acute headache. J Emerg Med. 2004;26(2):145-50.
- Thomas MC, Musselman ME, Shewmaker J. Droperidol for the treatment of acute migraine headaches. Ann Pharmacother. 2015;49(2):233-40.
- Roldan CJ, Chambers KA, Paniagua L, et al. Randomized Controlled Double-blind Trial Comparing Haloperidol Combined With Conventional Therapy to Conventional Therapy Alone in Patients With Symptomatic Gastroparesis. Acad Emerg Med. 2017;24(11):1307-14.
- Ramirez R, Stalcup P, Croft B, et al. Haloperidol undermining gastroparesis symptoms (HUGS) in the emergency department. Am J Emerg Med. 2017;35(8):1118-20.
- Vinson DR, Drotts DL. Diphenhydramine for the prevention of akathisia induced by prochlorperazine: a randomized, controlled trial. Ann Emerg Med. 2001;37(2):125-31.
- D'Souza RS, Mercogliano C, Ojukwu E, et al. Effects of prophylactic anticholinergic medications to decrease extrapyramidal side effects in patients taking acute antiemetic drugs: a systematic review and meta-analysis. Emerg Med J. 2018;35(5):325-31.
- Mahmoud M, Mason KP. Dexmedetomidine: review, update, and future considerations of paediatric perioperative and periprocedural applications and limitations. Br J Anaesth. 2015;115(2):171-82.
- Tang C, Xia Z. Dexmedetomidine in perioperative acute pain management: a non-opioid adjuvant analgesic. J Pain Res. 2017;10:1899-904.
- Wilson C. Feeling blocked? Another pain management tool in the emergency department. Ann Emerg Med. 2018 Aug;72(2):120-126.
- Moore 2014, Wiffen PJ, Derry S, et al. Gabapentin for chronic neuropathic pain and fibromyalgia in adults. Cochrane Database Syst Rev. 2014;(4):CD007938.
- Stewart B, Kugler A, Thompson P, et al. A saturable transport mechanism in the intestinal absorption of gabapentin is the underlying cause of the lack of proportionality between increasing dose and drug levels in plasma. Pharm Res 1993;10:276–81
- Neurontin (gabapentin) [package insert]. New York, NY: Parke Davis; September 2015.
- Lyrica (pregabalin) [package insert]. New York, NY: Pfizer; June 2012.
- Obata H. Analgesic mechanisms of antidepressants for neuropathic pain. Int J Mol Sci. 2017;18(11).
- Moore RA, Derry S, Adlington D, et al. Amitriptyline for neuropathic pain in adults. Cochrane Database Syst Rev. 2015;(7):CD008242
- Stahl SM, Grady MM, Moret C, et al. SNRIs: their pharmacology, clinical efficacy, and tolerability in comparison with other classes of antidepressants. CNS Spectr. 2005;10(9):732–747.
- Obata H. Analgesic mechanisms of antidepressants for neuropathic pain. Int J Mol Sci. 2017 Nov; 18(11): 2483.
- Burstein R, Jakubowski M. Analgesic triptan action in an animal model of intracranial pain. Ann Neurol. 2004;55:27–36.
- Coulter DM, Passier JL, Clark DW, et al. Activation of pain by sumatriptan. Headache. 2003;43(9):994-999.
- Tepper SJ, Rapoport AM, Sheftell FD. Mechanisms of action of the 5-HT1B/1D receptor agonists. Arch Neurol. 2002;59(7):1084–1088.
- Durham P, Russo A. New insights into the molecular actions of serotonergic antimigraine drugs. Pharmacol Ther. 2002;94(1–2):77–92.
- Tfelt-Hansen P, De Vries P, Saxena PR. Triptans in migraine: A comparative review of pharmacology, pharmacokinetics, and efficacy. Drugs. 2000;50:1259–1287.
- Gobel H, Winter P, Boswell D, et al. Comparison of naratriptan and sumatriptan in recurrence-prone migraine patients. Clin Ther. 2000;22:981–989.
- Pascual J, Munoz P. Correlation between lipophilicity and triptan outcomes. Headache. 2005;45(1):3-6.
- Imitrex (sumatriptan), package insert. Research Triangle Park, NC: GlaxoSmithKline; 2006.
- Zomig ZMT. Nasal Spray (zolmitriptan), package insert. Wilmington, DE: AstraZeneca; 2006.
- Amerge (naratriptan), package insert. Research Triangle Park, NC: GlaxoSmithKline; 2006.
- Maxalt, Maxalt-MLT (rizatriptan), package insert. Whitehouse Station, NJ; Merck & Co; 2007.
- Axert (almotriptan), package insert. Raritan, NJ: Ortho-McNeil; 2006.
- Frova (frovatriptan), package insert. Chadds Ford, PA: Endo; 2005.
- Relpax (eletriptan), package insert. New York: Pfizer; 2006.
- Ryan RE. Patient treatment preferences and the 5-HT1B/1D agonists. Arch Intern Med. 2001;161:2545–2553.
- Rapoport AM, Tepper SJ, Sheftell FD, et al. Which triptan for which patient? Neurol Sci. 2006;27:S123–S129.
- Silberstein SD for the U.S. Headache Consortium. Practice Parameter: Evidence-Based Guidelines for Migraine Headache (An Evidence-Based Review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2000;55:754–762.
- Dahlof CG. Infrequent or non-response to oral sumatriptan does not predict response to other triptans: Review of four trials. Cephalalgia. 2006;26(2):98–106.
- Boothby LA, Doering PL, Hatton RC. Carisoprodol: a marginally effective skeletal muscle relaxant with serious abuse potential. Hosp Pharm. 2003;38:337–345.
- Witenko C, Moorman-Li R, Motycka C, et al. Considerations for the appropriate use of skeletal muscle relaxants for the management of acute low back pain. P T. 2014 Jun;39(6):427-35.
- Baclofen Tablet [package insert]. Sellersville, Pennsylvania: Teva Pharmaceuticals USA; June 2010.
- Valium (diazepam) [package insert]. Nutley, NJ: Roche Laboratories, Inc.; January 2008.
- Tizanidine (Zanaflex) tablets [package insert]. Hawthorne, NY: Acorda Therapeutics, Inc.; November 2013.
- Chou R, Peterson K, Helfand M. Comparative efficacy and safety of skeletal muscle relaxants for spasticity and musculoskeletal conditions: a systematic review. Journal of pain and symptom management. 2004;28(2):140-175.
- Van Tulder MW, Touray T, Furlan A, et al. Muscle relaxants for nonspecific low back pain: A systematic review within the framework of the Cochrane Collaboration. Spine. 2003;28(17):1978–1992.
- Van Tulder MW, Touray T, Furlan A, et al. Muscle relaxants for nonspecific low back pain: A systematic review within the framework of the Cochrane Collaboration. Spine. 2003;28(17):1978-1992.
- McCarberg B, D’Arcy Y. Options in topical therapies in the management of patients with acute pain. Postgrad Med. 2013;125 (4 suppl):19-24.
- Pasternak GW. Opioids and their receptors:Are we there yet? Neuropharmacology. 2014;76 PtB:198-203.
- Pathan H, Williams J. Basic opioid pharmacology: an update. Br J Pain. 2012; 6(1): 11–16.
- Compton P, Charuvastra VC, Ling W. Pain intolerance inopioid-maintained former opiate addicts: effect of long-acting maintenance agent. Drug Alcohol Depend. 2001:63(2):139-46.
- Alford DP, Compton P, Samet J. Acute Pain Management for Patients Receiving Maintenance Methadone or Buprenorphine Therapy. Ann Intern Med. 2006 ;144(2):127-134.
- Peng PW, Tumber PS, Gourlay D. Review article: perioperative pain management of patients on methadone therapy. Can J Anaesth. 2005;52(5):513-23.
- Fishman SM, Wilsey B, Mahajan G, et al. Methadone reincarnated: novel clinical applications with related concerns. Pain Med. 2002;3:330-48.
- Johnson RE, Fudala PJ, Payne R. Buprenorphine: considerations for pain management. J Pain Symptom Manage. 2005;29:297-326.
- Book SW, Myrick H, Malcolm R, et al. Buprenorphine for postoperative pain following general surgery in a buprenorphine-maintained patient. Am J Psychiatry. 2007;164(6):979.
- American College of Rheumatology Ad Hoc Group on Use of, Selective, and Drugs Nonselective Nonsteroidal Antiinflammatory. 2008. 'Recommendations for use of selective and nonselective nonsteroidal antiinflammatory drugs: an American College of Rheumatology white paper', Arthritis Rheum, 59: 1058-73.
- Bathum, L., E. Skjelbo, T. K. Mutabingwa, H. Madsen, M. Horder, and K. Brosen. 1999. 'Phenotypes and genotypes for CYP2D6 and CYP2C19 in a black Tanzanian population', Br J Clin Pharmacol, 48: 395-401.
- Benyamin, R., A. M. Trescot, S. Datta, R. Buenaventura, R. Adlaka, N. Sehgal, S. E. Glaser, and R. Vallejo. 2008. 'Opioid complications and side effects', Pain Physician, 11: S105-20.
- Bhasker, C. R., W. McKinnon, A. Stone, A. C. Lo, T. Kubota, T. Ishizaki, and J. O. Miners. 2000. 'Genetic polymorphism of UDP-glucuronosyltransferase 2B7 (UGT2B7) at amino acid 268: ethnic diversity of alleles and potential clinical significance', Pharmacogenetics, 10: 679-85.
- Black, R. A., and D. A. Hill. 2003. 'Over-the-counter medications in pregnancy', Am Fam Physician, 67: 2517-24.
- Burns, S. M., C. W. Cunningham, and S. L. Mercer. 2018. 'DARK Classics in Chemical Neuroscience: Fentanyl', ACS Chem Neurosci.
- Chang, A. K., P. E. Bijur, D. Esses, D. P. Barnaby, and J. Baer. 2017. 'Effect of a Single Dose of Oral Opioid and Nonopioid Analgesics on Acute Extremity Pain in the Emergency Department: A Randomized Clinical Trial', JAMA, 318: 1661-67.
- Christrup, L. L. 1997. 'Morphine metabolites', Acta Anaesthesiol Scand, 41: 116-22.
- Darbari, D. S., R. H. van Schaik, E. V. Capparelli, S. Rana, R. McCarter, and J. van den Anker. 2008. 'UGT2B7 promoter variant -840G>A contributes to the variability in hepatic clearance of morphine in patients with sickle cell disease', Am J Hematol, 83: 200-2.
- de Fays, L., K. Van Malderen, K. De Smet, J. Sawchik, V. Verlinden, J. Hamdani, J. M. Dogne, and B. Dan. 2015. 'Use of paracetamol during pregnancy and child neurological development', Dev Med Child Neurol, 57: 718-24.
- De Gregori, S., M. De Gregori, G. N. Ranzani, M. Allegri, C. Minella, and M. Regazzi. 2012. 'Morphine metabolism, transport and brain disposition', Metab Brain Dis, 27: 1-5.
- Dean, L. 2012. 'Codeine Therapy and CYP2D6 Genotype.' in V. Pratt, H. McLeod, L. Dean, A. Malheiro and W. Rubinstein (eds.), Medical Genetics Summaries (Bethesda (MD)).
- Dean, M. 2004. 'Opioids in renal failure and dialysis patients', J Pain Symptom Manage, 28: 497-504.
- Duguay, Y., C. Baar, F. Skorpen, and C. Guillemette. 2004. 'A novel functional polymorphism in the uridine diphosphate-glucuronosyltransferase 2B7 promoter with significant impact on promoter activity', Clin Pharmacol Ther, 75: 223-33.
- Eichelbaum, M., and B. Evert. 1996. 'Influence of pharmacogenetics on drug disposition and response', Clin Exp Pharmacol Physiol, 23: 983-5.
- Eichelbaum, M., and A. S. Gross. 1990. 'The genetic polymorphism of debrisoquine/sparteine metabolism--clinical aspects', Pharmacol Ther, 46: 377-94.
- Feierman, D. E., and J. M. Lasker. 1996. 'Metabolism of fentanyl, a synthetic opioid analgesic, by human liver microsomes. Role of CYP3A4', Drug Metab Dispos, 24: 932-9.
- Heiskanen, T., K. T. Olkkola, and E. Kalso. 1998. 'Effects of blocking CYP2D6 on the pharmacokinetics and pharmacodynamics of oxycodone', Clin Pharmacol Ther, 64: 603-11.
- Holzer, P. 2009. 'Opioid receptors in the gastrointestinal tract', Regul Pept, 155: 11-7.
- Hoskin, P. J., G. W. Hanks, G. W. Aherne, D. Chapman, P. Littleton, and J. Filshie. 1989. 'The bioavailability and pharmacokinetics of morphine after intravenous, oral and buccal administration in healthy volunteers', Br J Clin Pharmacol, 27: 499-505.
- Ishida, J. H., C. E. McCulloch, M. A. Steinman, B. A. Grimes, and K. L. Johansen. 2018. 'Opioid Analgesics and Adverse Outcomes among Hemodialysis Patients', Clin J Am Soc Nephrol, 13: 746-53.
- Kumar, K., and S. I. Singh. 2013. 'Neuraxial opioid-induced pruritus: An update', J Anaesthesiol Clin Pharmacol, 29: 303-7.
- LaPietra, Alexis, and Sergey Motov. 2019. 'A Country in Crisis: Opioid Sparing Solutions for Acute Pain Management', Accessed 116:2 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6461319/pdf/ms116_p0140.pdf.
- Lazaryan, M., C. Shasha-Zigelman, Z. Dagan, and M. Berkovitch. 2015. 'Codeine should not be prescribed for breastfeeding mothers or children under the age of 12', Acta Paediatr, 104: 550-6.
- Leppert, W., M. Malec-Milewska, R. Zajaczkowska, and J. Wordliczek. 2018. 'Transdermal and Topical Drug Administration in the Treatment of Pain', Molecules, 23.
- Makris, U. E., M. J. Kohler, and L. Fraenkel. 2010. 'Adverse effects of topical nonsteroidal antiinflammatory drugs in older adults with osteoarthritis: a systematic literature review', J Rheumatol, 37: 1236-43.
- Mayer, D. J., and D. D. Price. 1976. 'Central nervous system mechanisms of analgesia', Pain, 2: 379-404.
- Mazer-Amirshahi, M., S. Motov, and L. S. Nelson. 2018. 'Hydromorphone use for acute pain: Misconceptions, controversies, and risks', J Opioid Manag, 14: 61-71.
- McCarberg, B., and Y. D'Arcy. 2013. 'Options in topical therapies in the management of patients with acute pain', Postgrad Med, 125: 19-24.
- Monk, J. P., R. Beresford, and A. Ward. 1988. 'Sufentanil. A review of its pharmacological properties and therapeutic use', Drugs, 36: 286-313.
- Osborne, R., S. Joel, D. Trew, and M. Slevin. 1988. 'Analgesic activity of morphine-6-glucuronide', Lancet, 1: 828.
- Owen, J. A., D. S. Sitar, L. Berger, L. Brownell, P. C. Duke, and P. A. Mitenko. 1983. 'Age-related morphine kinetics', Clin Pharmacol Ther, 34: 364-8.
- Paul, D., K. M. Standifer, C. E. Inturrisi, and G. W. Pasternak. 1989. 'Pharmacological characterization of morphine-6 beta-glucuronide, a very potent morphine metabolite', J Pharmacol Exp Ther, 251: 477-83.
- Pham, P. C., K. Khaing, T. M. Sievers, P. M. Pham, J. M. Miller, S. V. Pham, P. A. Pham, and P. T. Pham. 2017. '2017 update on pain management in patients with chronic kidney disease', Clin Kidney J, 10: 688-97.
- Poklis, A. 1995. 'Fentanyl: a review for clinical and analytical toxicologists', J Toxicol Clin Toxicol, 33: 439-47.
- Rendic, S., and F. J. Di Carlo. 1997. 'Human cytochrome P450 enzymes: a status report summarizing their reactions, substrates, inducers, and inhibitors', Drug Metab Rev, 29: 413-580.
- Ruiz-Garcia, V., and E. Lopez-Briz. 2008. 'Morphine remains gold standard in breakthrough cancer pain', BMJ, 337: a3104.
- Smith, H. S. 2009. 'Opioid metabolism', Mayo Clin Proc, 84: 613-24.
- Smith, M. T. 2000. 'Neuroexcitatory effects of morphine and hydromorphone: evidence implicating the 3-glucuronide metabolites', Clin Exp Pharmacol Physiol, 27: 524-8.
- Trescot, A. M., S. Datta, M. Lee, and H. Hansen. 2008. 'Opioid pharmacology', Pain Physician, 11: S133-53.
- Vallejo, R., R. L. Barkin, and V. C. Wang. 2011. 'Pharmacology of opioids in the treatment of chronic pain syndromes', Pain Physician, 14: E343-60.
- Wood, J. D., and J. J. Galligan. 2004. 'Function of opioids in the enteric nervous system', Neurogastroenterol Motil, 16 Suppl 2: 17-28.
- Jamieson DG. The safety of triptans in the treatment of patients with migraines. Am J Med. 2002;112(2):135-140.
