



Antidysrhythmic drugs are broadly categorized into Vaughan-Williams classifications, and their effects should be carefully noted.
Case
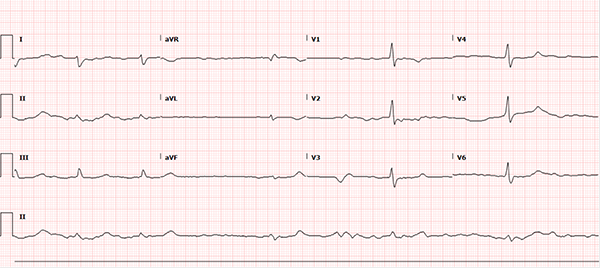
A 60-year-old female with a past medical history notable for atrial fibrillation, chronic alcoholism, and hypertension presents to the ED with 4 days of nausea and vomiting progressing to full-body weakness and malaise on the day of presentation. Upon arrival, the patient was hypotensive with a wide-complex, sinusoidal pattern noted on the monitor. The patient received calcium gluconate emergently for presumed hyperkalemia with subsequent narrowing of the QRS complex, after which the usual therapy for hyperkalemia was initiated. Additional history revealed the patient continued to take her home benazepril and metoprolol despite poor oral intake and gastrointestinal losses. She was recently started on flecainide by her cardiologist for atrial fibrillation one week prior. A repeat ECG, shown below, was obtained after the treatment above. What other etiologies should be considered in this patient?
Discussion
Electrocardiogram Case Discussion
This ECG rhythm is interpreted as atrial fibrillation with a slow ventricular response. The heart rate is irregularly irregular with a rate of approximately 36 beats per minute (BPM). There is no measurable PR interval secondary to the patient's underlying atrial fibrillation, but the QRS complex is widened at 140 milliseconds (ms). The corrected QT (QTc) interval for the patient’s heart rate is prolonged at approximately 490 ms. ST segment duration is 240 ms. ST segments and T-waves are unremarkable without clear ischemic changes and unremarkable morphology.
The Cardiac Action Potential and the Electrocardiogram
Although an in-depth discussion of the cardiac action potential is beyond the scope of this paper, a review of the physiology of cardiac myocyte conduction aids in the understanding of the ECG above and will be discussed in context. The cardiac action potential is divided into 5 sequential phases: Phase 0, Phase 1, Phase 2, Phase 3, and Phase 4. The healthy cardiac myocyte maintains a resting negative electrical potential (Phase 4) by an electrochemical gradient of ions. Intricately regulated ion transporters and channels allow passage through the relatively impermeable cellular membrane and coordinate the action potentials of cardiac myocytes. The action potential begins with a rapid influx of sodium ions triggering a rapid depolarization (Phase 0) of the cardiac myocyte. Once these sodium channels inactivate, there is a brief efflux of potassium from the cell causing a rapid, self-limited repolarization or "overshoot" (Phase 1).1,2
On the ECG, the QRS complex primarily reflects Phase 0 and, to a lesser extent, Phase 1 of cardiac myocyte depolarization across the surface of the heart.1 A normal QRS duration is between 60 and 120 ms. Widening of the QRS complex can be due to a myriad of causes including conduction system disease, electrolyte abnormalities, hypothermia, or blockade of the fast-acting sodium channel during Phase 0 of the cardiac action potential delaying the depolarization. Common culprits for the latter involve antidysrhythmic medications, cyclic antidepressants, phenothiazines, amantadine, diphenhydramine, carbamazepine and cocaine.3 In this case, the QRS remained prolonged despite normalization of extracellular potassium and stabilization of the cardiac membrane, and given the patient's history, this was suspicious for flecainide toxicity.
The plateau (Phase 2) of the action potential is orchestrated by sustained changes in the permeabilities of potassium and calcium ions through the cell membrane via numerous mechanisms. Finally, a delayed outward current caused by a rapid potassium efflux results in repolarization of the cell (Phase 3) with an eventual return to the Phase 4 resting potential. The ST segment on the ECG reflects the sustained depolarization (Phase 3) and the T-wave represents repolarization of the myocardium (Phase 4).1 The myocardium is susceptible to ventricular dysrhythmias if an unexpected depolarization occurs during this time. The QT interval varies with heart rate and is prolonged with bradycardia and may be corrected mathematically (QTc), yet remains prolonged in this case. The QT interval reflects ventricular systole and truly is an incorporation of multiple electrophysiological periods but in a simple model is primarily affected by increasing the duration of Phase 2 or 3 of the action potential. The factors are numerous but include physiologic modification, medication effects, genetic channelopathies or electrolyte abnormalities.3
Antidysrhythmic Therapy and Its Effects
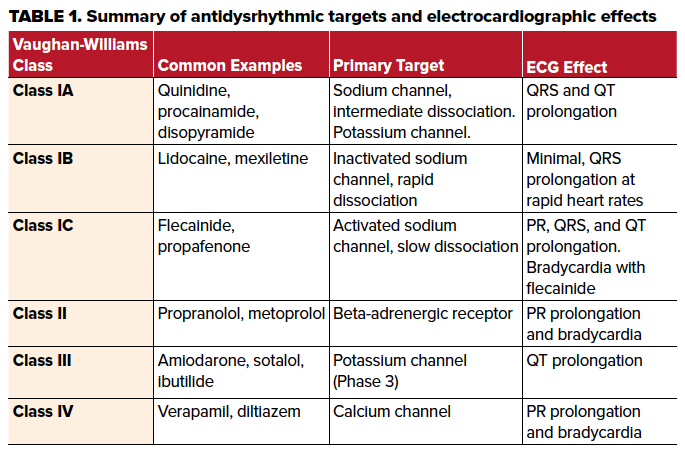
Broadly, antidysrhythmic therapies can be classified by the Vaughan-Williams classification system by the primary effect of each class on different targets in the heart.4 The major groups are labeled as classes I, II, III and IV with class I medications further subdivided into class IA, 1B and 1C based on additional electrophysiologic effects.2 It is important to note that this classification is far from perfect and there is significant cross-reactivity towards receptors targeted by other classes. Class I medications are defined primarily by their ability to block the rapid inward sodium current which defines Phase 0.2,4 Class IC medications, which include flecainide and propafenone, have the slowest dissociation from the activated receptor.4,5 Class IB medications, which include lidocaine and mexiletine, have the most rapid dissociation from the receptor and also preferentially bind to the inactivated or “closed” form of the channel which predominates at the end of Phase 0. In contrast to class IC medications, this causes electrocardiographic effects typically only seen at faster heart rates by a mechanism termed "use-dependence."5 Class IA medications such as procainamide, quinidine and disopyramide, have intermediate pharmacologic properties between class IB and class IC. Additionally, class IA medications exert an effect on the outward potassium channels in Phase 3 as well.4,5
As alluded to previously, the electrocardiographic effects of class I medications manifest with prolongation of the QRS complex via the mechanism above, along with other sodium channel blocking medications.2,5,6 With xenobiotics that affect sodium channel conduction, the right bundle of the cardiac conduction system is preferentially affected resulting in a delay of right ventricular activation. The associated ECG findings include an R wave in the terminal portion of the QRS complex in lead aVR and an S wave in leads 1 and aVL.3 The prolongation of the QRS complex by sodium channel blockade lengthens the QT interval, however it does not affect the ST interval duration as the ST interval generally reflects the duration of potassium efflux in Phases 2 and 3. This is in contrast to potassium channel blockade causes prolongation of both the QT interval and the ST segment duration.3
The medication in this case, flecainide, is a class IC medication indicated in the restoration of sinus rhythm in atrial fibrillation and the suppression of supraventricular tachycardia in structurally normal hearts.5 As a class IC antidysrhythmic, it primarily binds to activated sodium channel receptors with a slow dissociation, and there is likely some blockade of potassium channels as well.4,5 Flecainide’s strong affinity to activated sodium channels with slow dissociation leads to greater efficacy but also greater risk of adverse effects.5 Prolongation of the QT interval may lead to dangerous dysrhythmias such as ventricular fibrillation. As well, flecainide toxicity can also cause bradycardia.2,5 If P waves are present, flecainide may also prolong the PR interval on the ECG. Toxicity is suggested with a 50% increase in QRS duration, 30% prolongation of the PR interval or 15% prolongation of QTc interval.5,6 Flecainide also causes decreased inotropy which can lead to hypotension.5
Class II medications, which include beta-blockers, are defined as antagonists of the beta-adrenergic receptor. There are numerous examples of this class of antidysrhythmic. The beta-adrenergic receptor does not contribute to the cardiac myocyte action potential, but it aids in modulating heart rate primarily via effects on the conductivity of the atrioventricular (AV) node.1 Electrocardiographically, this is reflected by a slowing of the ventricular response and lengthening of the PR interval. In the case above, the patient was also prescribed metoprolol which likely contributed to the AV nodal blockade (ie, the slow ventricular response) along with the flecainide induced bradycardia. In general, class II antidysrhythmic medications have a minimal effect on the QRS interval or QT interval electrocardiographically.4
Class IV medications, which include verapamil and diltiazem, cause similar electrocardiographic changes as class II medications. These medications block slow, inward calcium channels in specialized pacemaker cells, which slow AV node conduction causing prolonged PR intervals, nodal blocks, and bradycardia with minimal effect on the QRS or QT duration.4
Class III medications, which include sotalol, ibutilide, and amiodarone, exert their effect at the outward rectifying potassium current responsible for repolarization in phase 3. This is reflected electrocardiographically as an increase in the QT interval and ST segment.4 It is important to note that although the Vaughan-Williams classification system attempts to neatly classify these medications, the specific compounds or their active metabolites may share mechanisms seen in other classes.4
Management
This discussion will focus primarily on class IC toxicity as seen in this case. Flecainide is renally excreted and excretion is improved with urinary acidification. As flecainide is a potent sodium channel inhibitor, hypertonic sodium bicarbonate may be useful in mitigating the widening of the QRS complex. Continued sodium bicarbonate infusions can cause alkalization of the urine so there is a theoretical benefit to utilizing sodium chloride infusion instead. Of note, there is no benefit for hemodialysis in flecainide toxicity.5 The patient in this case was also on a class II medication, which can cause bradycardia and AV nodal blockade. Atropine can be trialed initially, and intravenous glucagon and calcium may be pursued for more severe cases. High-dose insulin euglycemia protocols can also be effective. Hypotension due to beta-blocker toxicity is preferentially treated with catecholamine vasopressors followed by phosphodiesterase inhibitors.7 Intralipid therapy may be useful in both class IC and class II toxicities. Ultimately, in both cases, refractory hypotension and shock can be temporized with cardiopulmonary bypass or extracorporeal membrane oxygenation.5,7
Case Conclusion
This patient was admitted to the Medical Intensive Care Unit with a diagnosis of acute renal failure secondary to hypovolemia secondary to suspected alcoholic pancreatitis. The patient's hypotension, thought to be due to flecainide and metoprolol toxicity, was treated with norepinephrine and epinephrine infusions, aggressive volume resuscitation, and intralipid therapy. She was also given hypertonic bicarbonate with improvement in the duration of the QRS complex. The patient was ultimately discharged in stable and improved condition with normalization of her renal function and resolution of the abnormal findings seen on her initial ECG.
References
- Characteristics of Cardiac Muscle Cells. In: Mohrman DE, Heller L. eds. Cardiovascular Physiology. 9th ed. New York, NY: McGrawHill.
- Knollmann BC, Roden DM. Antiarrhythmic Drugs. In: Brunton LL, Hilal-Dandan R, Knollmann BC. eds. Goodman & Gilman's: The Pharmacological Basis of Therapeutics. 13th ed. New York, NY: McGraw-Hill.
- Clancy C. Electrophysiologic and Electrocardiographic Principles. In: Hoffman RS, Howland M, Lewin NA, Nelson LS, Goldfrank LR. eds. Goldfrank's Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill; 2015.
- Kowey PR. Pharmacological Effects of Antiarrhythmic Drugs. Arch Internal Med. 1998;158(4):325.
- Nelson LS. Antidysrhythmics. In: Hoffman RS, Howland M, Lewin NA, Nelson LS, Goldfrank LR. eds. Goldfrank's Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill; 2015.
- Levis J. ECG Diagnosis: Flecainide Toxicity. Perm J. 2012;16(4):53.
- Brubacher JR. β-Adrenergic Antagonists. In: Hoffman RS, Howland M, Lewin NA, Nelson LS, Goldfrank LR. eds. Goldfrank's Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill;2015.