



A young woman presents with a 3-week history of fatigue, nausea, vomiting, heavy bleeding, and lightheadedness. Within hours she is intubated in the ICU. Follow the case management to understand an atypical presentation of her condition - and the importance of treatment without delays.
An 18-year-old African American female presented to the ED complaining of a 3-week history of generalized fatigue, nausea, vomiting, heavy vaginal bleeding, lightheadedness. On the day prior to arrival in the ED, she developed progressively worsening dyspnea with exertion. She had been to several urgent care centers and EDs without receiving a diagnosis. She denied any fever, rash, diarrhea, or hematochezia. She denied alcohol, cigarettes, or other drug use and was not taking any medications. On presentation, her VS were as follows: 98.6 T, 140 HR, 22 RR, 180/90 mmHg BP, 96% oxygen saturation on room air. The patient appeared tired with pale conjunctiva. There was no rash or evidence of active bleeding. Pelvic exam showed minimal blood at the cervical os, and rectal exam showed normal brown stool in the rectal vault, which was hemoccult negative.
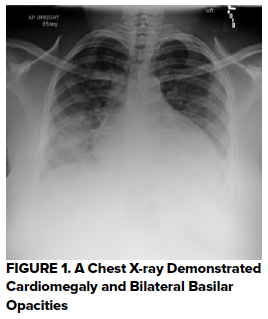
Initial lab work demonstrated WBC 23.6, Hb 5.1, HCT 14.9. The platelet count was indeterminate due to clumping. All other labs were marked as hemolyzed. Two units pRBCs were ordered for critical anemia. A bedside echo showed no pericardial effusion, no right heart strain, and normal ejection fraction. A FAST exam was negative for intraperitoneal fluid. A chest x-ray was obtained (Figure 1) with a radiology interpretation reporting, “Cardiomegaly with bilateral basilar opacities concerning bilateral pneumonia vs pulmonary hemorrhage.”
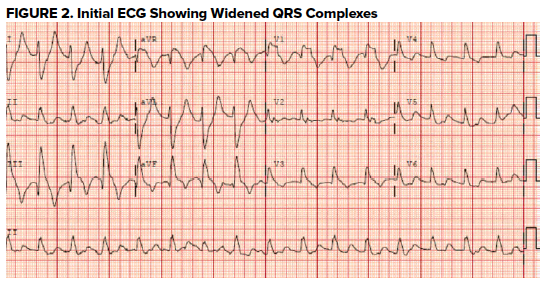
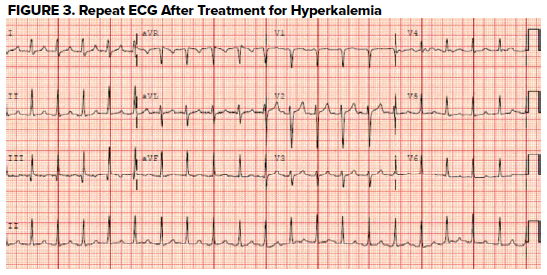
An ECG was obtained (Figure 2) showing a wide-complex rhythm with peaked T-waves. While awaiting BMP results, the patient was empirically treated for hyperkalemia with 3 g calcium gluconate, 1 amp sodium bicarbonate, 10 U insulin, 25 g D50, and nebulized albuterol. The QRS immediately narrowed on telemetry and on a subsequent ECG (Figure 3) that was obtained.
Results of a CBC with manual differential showed a platelet count of 75 and 2+ RBC fragments. Hemolysis labs and DIC screen were ordered. The patient's repeat BMP was reported as hemolyzed once again.
Given the clinical scenario of anemia with evidence of hemolysis, thrombocytopenia, and presumed hyperkalemia likely due to acute renal failure, thrombotic microangiopathy was considered with differential diagnoses including thrombotic thrombocytopenic purpura, hemolytic uremic syndrome, or complement-mediated hemolytic uremic syndrome. Nephrology and oncology were consulted for emergent dialysis and plasma exchange therapy (PLEX). A bedside istat was drawn showing a potassium of 7.5 and immeasurable creatinine. Telemetry showed recurrence of QRS widening requiring additional treatment for hyperkalemia, with improvement. The decision was made to place an emergent UDALL catheter to initiate dialysis and PLEX. After dialysis was started, the remainder of the laboratory studies results were:
- BMP: Na 131, K 9.4, Cl 85, CO2 6, BUN >186, Creatinine 48.3, Glucose 117, (Anion gap 39.6)
- Path review of smear: Moderate schistocytes
- Total bilirubin 0.3; LDH 1,328; haptoglobin < 10. PT; pTT and fibrinogen were within normal limits
All are consistent with microangiopathic hemolytic anemia.
The patient was admitted to the medical intensive care unit, where she required endotracheal intubation for acute hypoxic respiratory failure. Four hours of dialysis were completed with transfusion of 2 units of packed RBCs, and shortly afterwards PLEX was started.
Discussion
Microangiopathic hemolytic anemia (MAHA) is a condition defined by microvascular hemolysis with anemia and schistocyte formation. Thrombotic microangiopathy (TMA) is a condition characterized by MAHA and thrombocytopenia due to platelet activation and consumption. End-organ damage is caused by microvascular thrombi and occlusion leading to tissue ischemia.1,2
The major types of TMA are shiga toxin-mediated hemolytic uremic syndrome (ST-HUS, also known as classic HUS), complement-mediated TMA (also known as atypical HUS or aHUS), and thrombotic thrombocytopenic purpura (TTP). TMA is a hematologic emergency requiring prompt diagnosis and treatment. It should be suspected in any patient with evidence of hemolytic anemia and thrombocytopenia. The initial treatment of TMA includes supportive care, corticosteroids, and antibody removal with plasma exchange or plasmapheresis. Treatment should be started before definitive diagnosis is made, which can take several days.
Comparing TTP, ST-HUS, complement-mediated TMA
TTP is caused by decreased activity of ADAMTS13, most commonly due to inhibitory autoantibodies against ADAMTS13. ADAMTS13 is a protease that cleaves von Willebrand Factor (vWF) from large multimers to shorter molecules. Normally, endothelial cells produce long chains of vWF, and ADAMTS13 binds to cleavage points and cleaves the long molecules into shorter fragments. Without ADAMTS13, large chains of vWF accumulate in arterioles and capillaries, subsequently causing platelets to clump onto vWF, leading to microangiopathic occlusion causing end-organ damage.3 TTP is most commonly seen in young women. The classic pentad of TTP (found in only 5% of patients) is fever, microangiopathic hemolytic anemia, thrombocytopenia, renal failure, and neurologic symptoms (FAT RN). The hallmark of TTP is TMA with a severely reduced ADAMTS13 level of <10%.3
As previously mentioned, treatment with PLEX should not be delayed, as 50% of deaths from TTP occur in the first 24 hours.2 In cases where access to PLEX is delayed, FFP can be started, as it contains ADAMTS13, but PLEX has been shown to improve survival over FFP infusion alone.4 An ADAMTS13 level should be drawn before the initiation of PLEX or FFP to avoid a false-negative result.
ST-HUS is caused by shiga toxin, commonly produced by enterohemorrhagic Escherichia coli (EHEC) (usually O157:H7) and is associated with diarrhea. It is diagnosed by the identification of the shiga toxin or EHEC in the stool. Treatment with antibiotics may worsen the disease, and ST-HUS usually resolves with supportive care such as fluid resuscitation and blood transfusion.5
Complement-mediated TMA is an autoimmune-mediated HUS caused by complement dysregulation. The diagnosis is suspected in any patient with TMA with a normal ADAMTS13 level and negative stool studies. Compared to patients with TTP, patients with complement-mediated TMA are more likely to have severe renal dysfunction and pulmonary involvement (eg, pulmonary hemorrhage) and are less likely to have severe neurological changes. Prognosis of complement-mediated TMA is highly variable depending on which complement regulatory protein is mutated.6 Eculizumab is a monoclonal antibody used in the treatment for complement-mediated TMA. Eculizumab binds to the complement protein C5 and prevents conversion into C5a and C5b, ultimately preventing the production of membrane attack complex (MAC) and protecting RBCs from intravascular hemolysis.7,8
Conclusion
In the ICU, our patient underwent daily hemodialysis and PLEX with clinical and biochemical improvement and was extubated on hospital day 3. Workup revealed an ADAMTS13 antibody activity level within normal limits at 78%. Stool shiga toxin PCR was negative. She was found to have a decreased C3 level at 65 mg/dL (reference range 83-177) and was diagnosed with complement-mediated TMA. She was started on eculizumab and discharged from the hospital with outpatient hemodialysis.
Our patient presented with complement-mediated TMA with a symptom complex of acute renal failure, hemolytic anemia, and pulmonary hemorrhage. Due to delays in her initial diagnosis, she presented in an advanced stage of renal failure with life-threatening hyperkalemia.
In cases of TMA, the laboratory may delay reporting results due to hemolysis being assumed to be in vitro while in fact, the hemolysis is in vivo. This resulted in a delay in reporting both the CBC as well as the BMP results. Nonetheless, clinical care was not delayed as the EKG changes of hyperkalemia were recognized and promptly treated.
While the specific treatment of complement-mediated TMA centers on eculizumab, initial treatment for the TMAs is usually started with steroids and PLEX while awaiting further diagnostic results such as shiga toxin and ADAMTS13 levels.
References
- Tsai HM. Untying the knot of thrombotic thrombocytopenic purpura and atypical hemolytic uremic syndrome. Am J Med. 2013;126(3):200-209.
- Arnold DM, Patriquin CJ, Nazy I. Thrombotic microangiopathies: a general approach to diagnosis and management. CMAJ. 2017;189(4):E153-E159.
- Matsumoto M, Fujimura Y, Wada H, et al. Diagnostic and treatment guidelines for thrombotic thrombocytopenic purpura (TTP) 2017 in Japan. Int J Hematol. 2017;106(1):3-15.
- Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N Engl J Med. 1991;325(6):393-397.
- Brandt JR, Fouser LS, Watkins SL, et al. Escherichia coli O 157:H7-associated hemolytic-uremic syndrome after ingestion of contaminated hamburgers. J Pediatr. 1994;125(4):519-526.
- Caprioli J, Noris M, Brioschi S, et al. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood. 2006;108(4):1267-1279.
- Brodsky RA. Eculizumab: another breakthrough. Blood. 2017;129(8):922-923.
- Menne J, Delmas Y, Fakhouri F, et al. Outcomes in patients with atypical hemolytic uremic syndrome treated with eculizumab in a long-term observational study. BMC Nephrol. 2019;20(1):125.