Hepatorenal and hepatopulmonary syndromes can be easy to diagnose in the ED, and proper management can significantly impact morbidity and mortality downstream.
One of the top 10 leading causes of death in the United States, cirrhosis is defined as the end result of progressive hepatic fibrosis, most commonly due to infection (viral hepatitis) or long-term alcohol use. Cirrhosis is characterized by the progressive destruction of the underlying hepatic architecture, followed by the eventual loss of intrinsic function and an increase in portal venous pressures. Chronic increases in portal pressures lead to the majority of emergent complications due to cirrhosis, such as variceal bleeding, refractory ascites, hepatic encephalopathy, and spontaneous bacterial peritonitis (SBP). These conditions are frequently the cause for ED presentation, and standardized management guidelines are well-established.
While many of these complications are widely known and frequently recognized in the ED, hepatic dysfunction and increasing portal pressures can have additional effects on several other organ systems. These interactions, namely the hepatorenal and hepatopulmonary syndromes, can be easily diagnosed by initial clinical exam and laboratory findings. Prompt recognition and treatment of these conditions from within the ED can have a profound downstream impact on morbidity and mortality. This article will review these conditions and highlight key management strategies to streamline and improve downstream evaluation and care.
Hepatorenal Syndrome
Hepatorenal syndrome (HRS), characterized by renal failure and major impairment in circulatory function, is a common complication of advanced cirrhosis and fulminant liver failure. In one prospective study of 229 patients with cirrhosis complicated by ascites without elevation in their BUN, 18% had HRS at 1 year, and 39% had HRS at 5 years. The pathophysiology of HRS is defined by the derangements of vasoconstriction and vasodilation that occur in advanced cirrhosis. Increased intrahepatic resistance and portal hypertension leads to an increased production of vasodilators within the splanchnic circulation, and subsequent dilation of both the splanchnic and systemic circulation as well as a low effective circulating blood volume. These effects cumulatively result in activation of the renin-angiotension-aldosterone system, increased cardiac output, and intense renal vasoconstriction, leading to renal failure and sodium retention, both hallmarks of HRS.
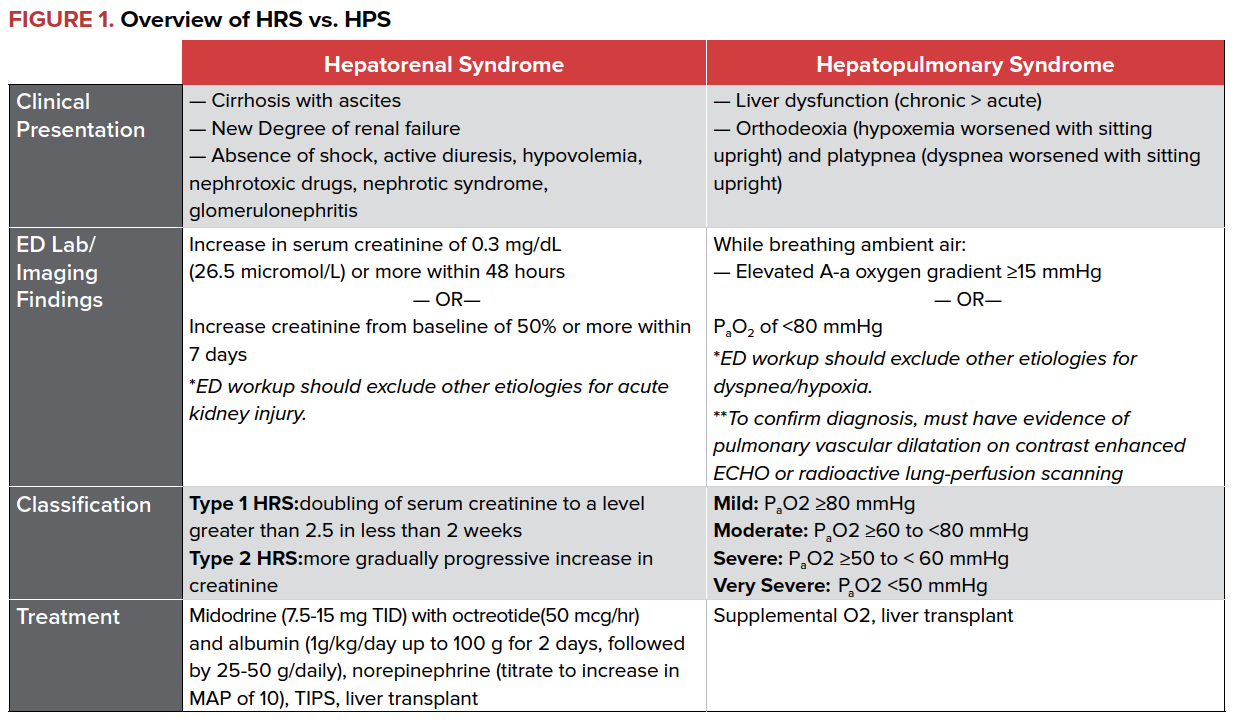
There are two types of HRS, classified based on the onset of renal failure. Type 1 HRS is characterized by rapid, progressive renal failure defined by a doubling of the serum creatinine to a level greater than 2.5 in less than 2 weeks and occurs as the result of a precipitant, such as SBP and/or GI bleeding. Type 2 HRS is a more gradual and less severe form of renal failure.
Diagnosis
Diagnosis of HRS can be challenging and is based on clinical findings. The ED clinician should consider HRS in patients presenting with stigmata of liver disease (spider angiomas, ascites, clubbing) and new or worsening renal failure as defined by rising creatinine and blood urea nitrogen (BUN). Importantly, HRS is a diagnosis of exclusion and can only be made once other etiologies of renal failure have been ruled out. Workup should be broad to exclude other etiologies of renal failure including infection, systemic shock, use of nephrotoxic drugs, and presence of proteinuria or hematuria.
Treatment
Management of HRS focuses on reversal of kidney injury by improving renal blood flow through the use of vasopressors and albumin. In patients who do not require ICU level of care, midodrine, octreotide, and albumin have shown a mortality benefit and resolution of HRS. Terlipressin, a vasopressin analogue, has been shown to be effective in the treatment of HRS, but is currently not available in North America.
For patients who require ICU level care, norepinephrine has also been shown to improve survival. Transjugular intrahepatic portosystemic shunt (TIPS) may have a role in treating HRS, but data is limited and more studies are needed. Ultimately, liver transplant is the treatment of choice.
Hepatopulmonary Syndrome (HPS)
Hepatopulmonary Syndrome (HPS) is the triad of liver disease, hypoxemia, and evidence of intrapulmonary vasodilation. In patients with liver disease, intrapulmonary vasodilatation is qualitatively identified through contrast enhanced echocardiography (“bubble study”) where agitated saline is injected through a peripheral vein and identified in the left atrium within 3-6 cardiac cycles.
The severity of HPS is based on the degree of hypoxemia as measured by the partial pressure of arterial oxygen in patients with an elevated alveolar-arterial (A-a) oxygen gradient. The prevalence of HPS varies depending on study site, with a reported average of approximately 25% of patients with chronic liver disease developing the syndrome. A majority of patients with HPS have underlying chronic liver disease with cirrhosis and portal hypertension; however, the criteria for HPS can be met with acute liver failure and ischemic hepatitis as well as in patients without evidence of portal hypertension or cirrhosis. The severity of liver disease is not correlated to the development of HPS.
The underlying pathogenesis of HPS is not well understood. The prevailing theory is that liver disease increases the risk for bacterial translocation and endotoxin release, leading to both increased production and decreased clearance of pulmonary vasodilators such as nitric oxide. Inhibition of hypoxic pulmonary vasoconstriction due to dilatation of pulmonary precapillary and capillary vessels is a defining feature of HPS leading to ventilation-perfusion mismatch. Diffusion limitation and, in some cases, intrapulmonary shunting are other pathophysiologic changes that occur in HPS.
The ED clinician should consider HPS in patients presenting with stigmata of liver disease, dyspnea, and hypoxemia. Patients may experience increased dyspnea (platypnea) and worsened hypoxemia (orthodeoxia) upon sitting upright from the supine position. Workup should be broad and include arterial blood gas analysis, chest radiography, and consideration of other etiologies for an increased A-a gradient, including pneumonia, cardiogenic pulmonary edema, pulmonary embolism, atelectasis, and pleural effusion.
There is no medical treatment available for HPS at this time. Supplemental oxygen is used to maintain saturations until liver transplantation can be performed. Even after transplant, resolution of arterial hypoxemia can take months.
Conclusion
Despite improvements in recognition and optimization of medical management, the ultimate prognosis of HRS remains poor. In one series, patients with type 1 HRS had a median survival of 1 month, and type 2 patients had a median survival of 3 months if their MELD was >20, and 11 months if MELD was < 20. Mortality even after transplant is also known to be higher in patients who have developed both types of HRS. On the other hand, patients with HPS undergoing liver transplant have a similar 5-year mortality compared to individuals without HPS going into transplant.