



A 50-year-old male is brought in by EMS after pulseless electrical activity (PEA) cardiac arrest. Reportedly, he was well earlier in the day before family witnessed him collapse with seizure-like activity. Bystanders began CPR before EMS initiated care for PEA with subsequent ROSC. Post-arrest, the patient was hypotensive with a 12-lead electrocardiogram (EKG) notable for third degree heart block. On arrival to ED, he was being transcutaneously paced. Norepinephrine was started and a transvenous pacemaker was placed with stabilization of his hemodynamics. The patient was taken to CT and found to have a high-grade aneurysmal subarachnoid hemorrhage from a ruptured aneurysm of the anterior communicating artery.
How does subarachnoid hemorrhage lead to cardiac arrest, complete heart block, or cardiomyopathy? How frequently does this occur? What is neurogenic stress cardiomyopathy? How does it threaten the neurologically injured patient, and what implications does it have on their care?
Introduction
During aneurysmal subarachnoid hemorrhage (aSAH), extravasation of high-pressure arterial blood into the subarachnoid space leads to a sudden intracranial pressure (ICP) increase that can compromise cerebral perfusion. Blood is most commonly found around the circle of Willis (~65%) due to the proximity of the majority of aneurysms arising within this region, or in the Sylvian fissure (~30%).1 Within the Sylvian fissure underlying the cortex lie the insular lobe and deeper hypothalamus which have important clinical implications, to be discussed later. (Figure 1)
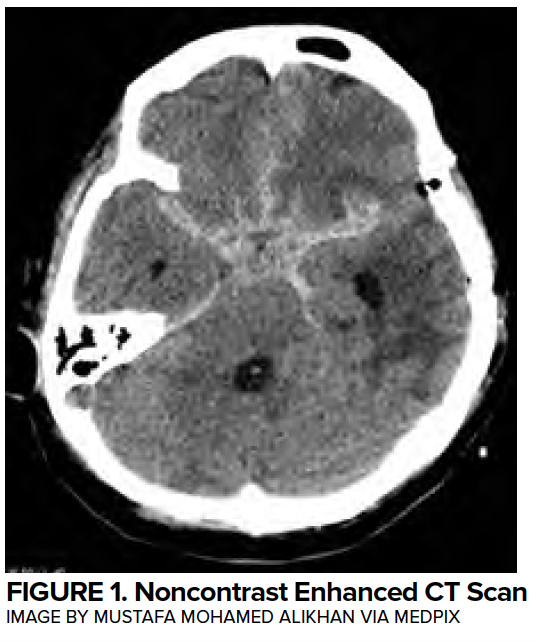
A subset of subarachnoid hemorrhage patients present after cardiac arrest, often in PEA. Among all PEA arrest cases, approximately 7% are due to intracranial hemorrhage.2 In distinction from other stroke patients,3 aSAH occur in younger patients without cardiovascular risk factors. An exploration into neurocardiology—the unique interface between the central nervous and cardiovascular systems—will help explain how a neurologic insult such as non-traumatic aneurysmal subarachnoid hemorrhage (aSAH) can lead to acute cardiac dysfunction.
Cardiac Manifestations
Myocardial injury commonly occurs following aSAH, with 28% of patients with troponin elevations in the first 24 hours,4 35% with arrhythmias,5 and 28% with wall motion abnormalities.6 Classically, deep septal T-wave inversions (cerebral T waves) and QTC prolongation (associated with insular injury) can be found on EKG. (Figure 2)
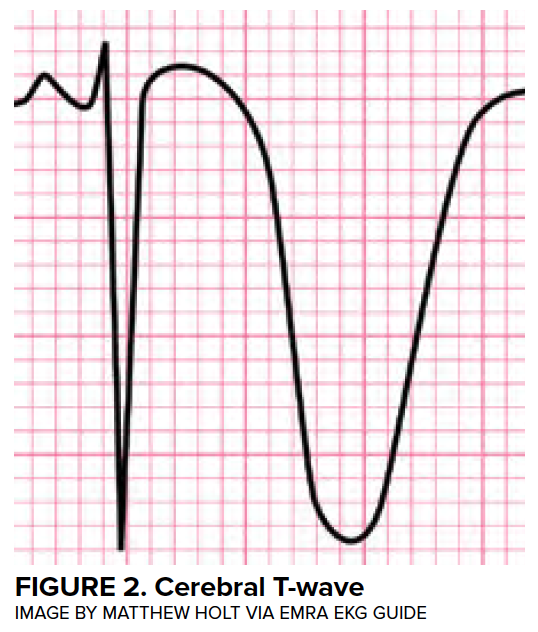
While almost all patients will have some abnormality on EKG, and the degree of abnormality is proportional to the cerebral insult,7 a wide range of findings can be observed. Life-threatening arrhythmias are seen in just 5% of cases.5
The degree of troponin elevation correlates with both intracranial injury,8 left ventricular (LV) dysfunction, and mortality.9,10 Regional wall motion abnormalities (RWMA) WMAs extending beyond single epicardial distribution hint at the underlying pathophysiology (to be discussed below), and occur more frequently among postmenopausal women with aSAH.
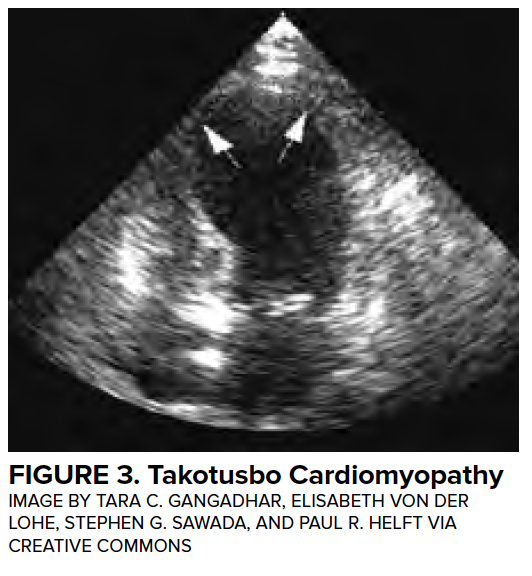
Transient LV dysfunction in the setting of neurologic injury is commonly described as “neurogenic heart” or “neurologic stunned myocardium” and will be referred to as neurogenic stress cardiomyopathy (NSCM) in this article. Many other disease entities have been described to cause a characteristic, self-resolving LV dysfunction, and the medical literature describes more than 70 different terms.11 Definitions of neurogenic stress cardiomyopathy vary but often require the both reversible or transient LV dysfunction to be reversible and a negative coronary angiogram. Estimates from retrospective studies suggest that the incidence of neurogenic stress cardiomyopathy in patients with aSAH range from 1-5%,12,13 although these studies likely underestimate the true prevalence. Importantly, LV dysfunction is associated with high-grade SAH and is associated with a mortality of 26%.12 (Figure 3)
Pathophysiology
NSCM was thought to be caused by myocardial ischemia or coronary vasospasm in response to a surge in adrenal circulating catecholamines, but more likely is directly related to myocardial sympathetic stimulation by a damaged insular cortex.14-16 This results in myocardial contraction, adenosine triphosphate depletion, mitochondrial dysfunction, and myocardial cell dysfunction or death.17 The typical apical and mid-ventricular ballooning pattern of neurogenic stress cardiomyopathy reflects the differential distribution of myocardial sympathetic nerve terminals.18 The subendocardial myocardium receiving autonomic innervation also houses the cardiac conduction system, predisposing injured subendocardial myocytes to arrhythmogenicity.
Management
Emergent care for the aSAH patient has two concurrent priorities:
- Obliteration of the aneurysm.
- Optimizing the hemodynamics and cerebral perfusion to avoid secondary injury.
Prior to aneurysmal repair, the latter entails avoiding hypertension (SBP<160) to decrease risk of rebleeding, the most feared early complication. Rebleeding has a 22% incidence, often occurs in the first 6 hours,19 and is associated with 67% mortality.20 Cerebral autoregulation— response of cerebral vasculature to changes in systemic blood pressure by altering vascular tone in order to ensure steady perfusion—is impaired in the first 24 hours (or beyond) in the neurologically injured patient, so the brain is exquisitely sensitive to fluctuations in blood pressure.
The cornerstone of treatment for patients with concurrent NSM and SAH is optimizing cardiac output to ensure adequate cerebral perfusion. Cardiac dysfunction is often early and transient (1-3 days);6 however, neurogenic stress cardiomyopathy can be prolonged for weeks. If vasopressor support is required, norepinephrine is a reasonable first line agent, and dobutamine may be added for additional inotropy. Rarely, mechanical circulatory support devices such as Impella21 or intraortic balloon pump22 are used.
In addition to potentially impaired cerebral perfusion due to depressed cardiac output, NSM presents several other management challenges. First, induction of anesthesia for aneurysm coiling or clipping procedures often results in further myocardial suppression, which can result in hemodynamic collapse in patients with severe NSM. Second, a major late complication of aSAH is vasospasm leading to delayed cerebral ischemia. It patients are unable to maintain adequate cerebral perfusion pressures in the face of high cerebrovascular resistance due to decreased cardiac output, cerebral ischemia will develop. These are patients who often require inotropic or mechanical support.
Case Conclusion and Takeaways
The patient had an external ventricular drain placed in the ED and was admitted to the neurological intensive care unit. He was taken for cerebral angiography and his aneurysm was coiled. He remained on a norepinephrine infusion to maintain adequate cerebral perfusion pressure. A permanent pacemaker was placed, and he was discharged with good neurologic function.
In summary, cardiovascular complications from aSAH are common and neurogenic stress cardiomyopathy can be severe. Emergency neurological life support (ENLS) recommends early screening for cardiac dysfunction, including obtaining an EKG and troponin within the first hour of ED presentation.23 Maintaining normal blood pressure is essential, as aSAH patients may lose the ability to autoregulate cerebral blood flow and are particularly vulnerable to secondary ischemic insults. Treatment is supportive and may require vasopressors, inotropes, or rarely mechanical support to ensure adequate cerebral perfusion.
References
1. Hacking C & Gaillard F. Subarachnoid haemorrhage. Available at: https://radiopaedia.org/articles/subarachnoid-haemorrhage
2. Beun L, Yersin B, Osterwalder J, Carron PN. Pulseless electrical activity cardiac arrest: time to amend the mnemonic "4H&4T"?. Swiss Med Wkly. 2015;145:w14178.
3. Gongora-rivera F, Labreuche J, Jaramillo A, Steg PG, Hauw JJ, Amarenco P. Autopsy prevalence of coronary atherosclerosis in patients with fatal stroke. Stroke. 2007;38(4):1203-10.
4. Deibert E, Barzilai B, Braverman AC, et al. Clinical significance of elevated troponin I levels in patients with nontraumatic subarachnoid hemorrhage. J Neurosurg. 2003;98(4):741-6.
5. Solenski NJ, Haley EC, Kassell NF, et al. Medical complications of aneurysmal subarachnoid hemorrhage: a report of the multicenter, cooperative aneurysm study. Participants of the Multicenter Cooperative Aneurysm Study. Crit Care Med. 1995;23(6):1007-17.
6. Banki N, Kopelnik A, Tung P, et al. Prospective analysis of prevalence, distribution, and rate of recovery of left ventricular systolic dysfunction in patients with subarachnoid hemorrhage. J Neurosurg. 2006;105(1):15-20.
7. Brouwers PJ, Wijdicks EF, Hasan D, et al. Serial electrocardiographic recording in aneurysmal subarachnoid hemorrhage. Stroke. 1989;20(9):1162-7.
8. Tung P, Kopelnik A, Banki N, et al. Predictors of neurocardiogenic injury after subarachnoid hemorrhage. Stroke. 2004;35(2):548-51.
9. Naidech AM, Kreiter KT, Janjua N, et al. Cardiac troponin elevation, cardiovascular morbidity, and outcome after subarachnoid hemorrhage. Circulation. 2005;112(18):2851-6.
10. Vannemreddy P, Venkatesh P, Dinesh K, Reddy P, Nanda A. Myocardial dysfunction in subarachnoid hemorrhage: prognostication by echo cardiography and cardiac enzymes. A prospective study. Acta Neurochir Suppl. 2010;106:151-4.
11. Sharkey SW, Lesser JR, Maron MS, Maron BJ. Why not just call it takotsubo cardiomyopathy: a discussion of nomenclature. J Am Coll Cardiol. 2011;57(13):1496-7.
12. Abd TT, Hayek S, Cheng JW, Samuels OB, Wittstein IS, Lerakis S. Incidence and clinical characteristics of takotsubo cardiomyopathy post-aneurysmal subarachnoid hemorrhage. Int J Cardiol. 2014;176(3):1362-4.
13. Lee VH, Connolly HM, Fulgham JR, Manno EM, Brown RD, Wijdicks EF. Takotsubo cardiomyopathy in aneurysmal subarachnoid hemorrhage: an underappreciated ventricular dysfunction. J Neurosurg. 2006;105(2):264-70.
14. Mertes PM, Carteaux JP, Jaboin Y, et al. Estimation of myocardial interstitial norepinephrine release after brain death using cardiac microdialysis. Transplantation. 1994;57(3):371-7.
15. Nguyen H, Zaroff JG. Neurogenic stunned myocardium. Curr Neurol Neurosci Rep. 2009;9(6):486-91.
16. Kono T, Morita H, Kuroiwa T, Onaka H, Takatsuka H, Fujiwara A. Left ventricular wall motion abnormalities in patients with subarachnoid hemorrhage: neurogenic stunned myocardium. J Am Coll Cardiol. 1994;24(3):636-40.
17. Samuels MA. Neurogenic heart disease: a unifying hypothesis. Am J Cardiol. 1987;60(18):15J-19J.
18. Zaroff JG, Rordorf GA, Ogilvy CS, Picard MH. Regional patterns of left ventricular systolic dysfunction after subarachnoid hemorrhage: evidence for neurally mediated cardiac injury. J Am Soc Echocardiogr. 2000;13(8):774-9.
19. Inagawa T, Kamiya K, Ogasawara H, Yano T. Rebleeding of ruptured intracranial aneurysms in the acute stage. Surg Neurol. 1987;28(2):93-9.
20. Winn HR, Richardson AE, Jane JA. The long-term prognosis in untreated cerebral aneurysms: I. The incidence of late hemorrhage in cerebral aneurysm: a 10-year evaluation of 364 patients. Ann Neurol. 1977;1(4):358-70.
21. Rashed A, Won S, Saad M, Schreiber T. Use of the Impella 2.5 left ventricular assist device in a patient with cardiogenic shock secondary to takotsubo cardiomyopathy. BMJ Case Rep. 2015;2015.
22. Al-mufti F, Morris N, Lahiri S, et al. Use of Intra-aortic- Balloon Pump Counterpulsation in Patients with Symptomatic Vasospasm Following Subarachnoid Hemorrhage and Neurogenic Stress Cardiomyopathy. J Vasc Interv Neurol. 2016;9(1):28-34.
23. Edlow BL, Samuels O. Emergency Neurological Life Support: Subarachnoid Hemorrhage. Neurocrit Care. 2017;27(Suppl 1):116-123.