



A 49-year-old male with a history of alcohol abuse presents to the ED with complaints of generalized abdominal pain and vomiting for the last 36 hours. The patient is well-known to the department for alcohol-related visits and continues to drink daily. On arrival, he is tachycardic and tachypneic, and physical examination findings include dry mucous membranes, decreased sakin turgor, epigastric tenderness, and a tremor in both hands. Laboratory studies show a serum bicarbonate of 10 mEq/L, an anion gap of 30, a serum glucose of 95 mg/dL, a lactic acidosis with pH 7.2, hypophosphatemia, and trace ketonuria. Abdominal CT scan is normal. He denies a history of diabetes mellitus, ingestion of any toxic alcohols, or recent illness.
This patient could potentially have any one of many diagnoses, but his presentation and lab findings are most consistent with alcoholic ketoacidosis (AKA). AKA can be a common ED diagnosis and typically occurs in chronic alcohol drinkers who have an abrupt cessation in their alcohol intake coupled with decreased glycemic intake and intravascular volume depletion.1
In the majority of cases, a precipitating event such as pancreatitis, gastritis, or an aspiration pneumonia leads to an abrupt decline in oral intake. About 24 to 72 hours after cessation of PO intake, AKA can develop.2 These patients usually have a low or absent serum alcohol concentration and can present with varying degrees of alcohol withdrawal. However, a clear sensorium is a hallmark of this condition. The presence of an alteration in consciousness strongly suggests that another process is present.3
Although the underlying pathophysiology is complex, a proper comprehension greatly aids in the diagnosis and management of this condition.
There are three general concepts that drive AKA:
- Alcohol ingestion, compounded with decreased caloric intake and dehydration, favors a ketotic state.
Ketoacidosis is caused by a combination of factors, including starvation-induced hypoinsulinemia, oxidation of alcohol to its various ketone metabolites, lipolysis with free fatty acid (FFA) release, and intravascular volume contraction. The relative starvation state in AKA leads to excessive glucagon secretion and reduced peripheral insulin concentrations, which plays a key role in developing ketoacidosis. Metabolism of fats through lipolysis produces beta-hydroxybutyrate (BHB) and acytyl-acetate (ACA). These ketones are utilized for cellular respiration to provide energy through adenosine triphosphate (ATP) production, but add to the anion gap acidosis seen in AKA.
- During the metabolism of ethanol, high amounts of NADH (the reduced form of nicotinamide-adenine dinucleotide [NAD+]) are generated.4
NAD+ is a coenzyme used to carry electrons in intracellular redox reactions. The reduction of NAD+ and consequential accumulation and imbalance of NADH in the metabolism of ethanol has several important consequences. BHB generation predominates over the production of ACA in this high NADH to NAD+ ratio. This abnormal ratio leads to an inhibition of the citric acid cycle and hepatic gluconeogenesis, which partially explains why hyperglycemia is rare in these patients.Almost counterintuitively, there is a failure to regenerate normal levels of NAD+ and ACA in AKA. The reoxidization of NADH to NAD+ appears to be limited by a combination of factors, including hypophosphatemia and a functional block within the mitochondria.2The lactic acidosis seen in AKA is due to an abnormal redox state. Pyruvate is a substrate used in numerous energy-producing pathways, but in alcoholic ketoacidosis, it is shifted from its normal metabolic pathways to others that increase lactate production. In addition, the regeneration of pyruvate from lactic acid is impaired.
- A heightened adrenergic state and volume depletion worsen ketosis and inhibits gluconeogenesis, creating a state that favors the creation and maintenance of a ketotic milieu.
The body responds to starvation, dehydration, and hypoglycemia with the release of counter-regulatory hormones. These hormones increase sympathetic tone, decrease insulin release, and increase ketone concentration through the release of FFAs and decreased peripheral ketone metabolism. All of these changes perpetuate the ketotic state until glucose is reintroduced into the system. Significant dehydration due to vomiting and decreased oral intake lead to impaired renal ketone clearance, further exacerbating the situation.2The differential diagnosis for AKA should include starvation ketosis and diabetic ketoacidosis (DKA). Although a thorough history can help to narrow the differential, a metabolic panel is essential to confirm the diagnosis. Anion gaps of 30 mEq/L or more can be seen in AKA, though the gap may be obscured by a concomitant primary metabolic alkalosis due to vomiting. In fact, there are case reports of patients with AKA who have an alkalemic serum pH due to excessive vomiting.The anion gap in starvation ketosis is typically much lower, with bicarbonate levels rarely below 18 mEq/L, and serum pH typically above 7.30.2 In DKA, by contrast, the anion gap can be quite high, with bicarbonate levels frequently reaching the single digits. Hyperglycemia with glycosuria, typically seen in diabetic ketoacidosis (DKA), is rare with AKA.4 Chronic malnutrition leads to low glycogen reserves, and the heightened adrenergic tone leads to inhibition of hepatic gluconeogenesis. Ketonuria, present in all three of these conditions, can confound the severity of AKA.Ketonuria is measured by the nitroprusside test, in which a color change indicates the relative concentration of acetone and ACA in the urine. The presence of BHB, the most prominent ketone present in AKA, is not reflected by the nitroprusside test. This explains why patients with AKA may show no or only slight ketonuria on initial presentation, with a paradoxical increase as the condition is reversed. As the ACA:BHB ratio normalizes, both the detectable ACA and BHB are cleared in the urine.
Differential diagnosis
Other life-threatening conditions that can cause a significant anion gap acidosis should also be considered in the differential diagnosis. The toxic alcohols, specifically methanol and ethylene glycol, may be intentionally or accidentally ingested in this patient population. These ingestions can cause significant morbidity and mortality if not appropriately managed.5 Altered mental status is a common feature of toxic alcohol ingestion but is not usually seen in AKA.5
Patients will typically have an initial osmolar gap that transitions to an increased anion gap as the toxic alcohol is metabolized. Elevated serum BHB concentration may be quite elevated in AKA, but this does not necessarily exclude the possibility of toxic alcohol ingestion; nor does the absence of an osmolar or anion gap rule out the diagnosis. While patients in AKA have a slight lactic acidosis, the presence of a significantly elevated lactate level should prompt the search for an underlying illness. Rarely, a combination of AKA and one of these other events may occur and present a diagnostic conundrum. Thoughtful consideration of timing, type and amount of ingestion, and associated symptoms, in combination with observation and laboratory studies, must be used to make this differentiation if a clear and accurate history is lacking.
Treatment
The reversal of ketosis and vigorous rehydration are central in the management of AKA. In addition to isotonic fluid replacement, dextrose-containing intravenous fluids are needed. Typically, 5% dextrose with half-normal saline at a rate of 150 mL per hour provides sufficient glucose to stimulate the pancreas to secrete insulin, allowing peripheral tissues to metabolize ketones and inhibit FFA release.2 It also allows the body to regenerate NAD+, which is inhibited by the metabolic alterations caused by AKA. Intravenous dextrose-containing fluid infusions should be stopped once the bicarbonate levels have reached 18-20 mEq/L and the patient is tolerating oral intake. This typically occurs 8 to 16 hours after the initiation of treatment.2 Alcohol withdrawal in these patients should be aggressively managed with intravenous benzodiazepines. Thiamine, folate, and other electrolytes, most notably phosphate and potassium, may need to be repleted in these patients.6 Interestingly, the majority of morbidity seen in AKA is due to the underlying process that caused the cessation of alcohol.
Case conclusion
The patient received 4 liters of normal saline and was started on D5-1/2 NS prior to admission. He was given IV valium for alcohol withdrawal, and thiamine, folate, and phosphate were repleted. He was hospitalized for three days for management of AKA and alcohol withdrawal, then discharged once tolerating oral intake and in good condition. He was seen three weeks later in the emergency department for a similar presentation.
Table 1. Characteristics of Common Ketoacidoses
| Diabetic Ketoacidosis | Alcoholic Ketoacidosis | Starvation Ketoacidosis | |
| Bicarbonate | Can reach single digits | Can reach single digits | > 18 |
| Glucose | Elevated | Low to mildly elevated | Low to normal |
| Measurable ketonuria | Present | Absent or present | Present |
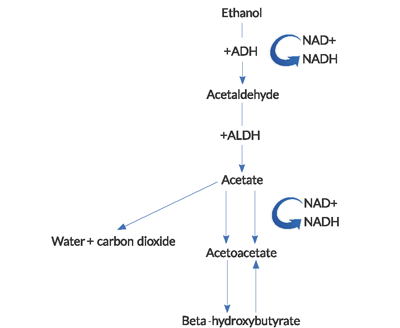
Figure 1. Pathway of alcohol metabolism
(ADH = alcohol dehydrogenase, ALDH = acetaldehyde dehydrogenase).
References
- Palmer, Jerry P. Alcoholic ketoacidosis: Clinical and laboratory presentation, pathophysiology and treatment. Clinics in endocrinology and metabolism 12.2 (1983): 381-389.
- Duffens K, Marx JA. Alcoholic ketoacidosisa review.The Journal of emergency medicine 5.5 (1987): 399-406.
- Wrenn KD, Slovis CM, Minion GE, et al. The syndrome of alcoholic ketoacidosis. The American journal of medicine 91.2 (1991): 119-128.
- Marx JA, Hockberger RS, Walls RM, et al., eds. Rosens™ Emergency Medicine: Concepts and Clinical Practice. Philadelphia, PA: Mosby/Elsevier; 2013. Chapter 185 Alcohol-Related Disease by John T. Finnell.
- Kraut JA, Kurtz I. “Toxic alcohol ingestions: clinical features, diagnosis, and management. Clinical Journal of the American Society of Nephrology 3.1 (2008): 208-225.
- Miller PD, Heinig R, Waterhouse C. Treatment of alcoholic acidosis: the role of dextrose and phosphorus. Archives of internal medicine 138.1 (1978): 67-72.