



Patients with alcohol use disorder commonly present to the ED critically ill, with myriad underlying pathologies. Alcoholic ketoacidosis (AKA) should be considered in anyone with prolonged and/or binge consumption of alcohol.
AKA is a diagnosis of exclusion, and many other life-threatening alternative or concomitant diagnoses present similarly, and must be ruled out. Failure to make the diagnosis can result in severe metabolic abnormalities, acidosis, and shock.
Case
A 32-year-old male presented to the ED with 24 hours of consistent non-bloody, non-bilious emesis after a 10-day alcohol binge. He reported nausea, vomiting, abdominal pain, chills, and sweats. His past medical history included alcoholic cardiomyopathy (left ventricular ejection fraction of 20%), renal artery embolism, and upper GI bleed of unknown etiology. The patient’s home medications include digoxin, sacubitril/valsartan, eplerenone, metoprolol, pantoprazole, and warfarin; however he reported medication noncompliance for 11 days prior to presentation. On exam, he was toxic appearing and in moderate distress secondary to pain. Vital signs were significant for a heart rate of 118 beats per minute and a blood pressure of 113/72. He was tremulous and had epigastric tenderness to palpation. The remainder of his physical exam was unremarkable. The differential diagnosis on initial evaluation was broad, and included pancreatitis, alcohol-induced gastritis, alcohol withdrawal, alcoholic hepatitis, sepsis, toxic alcohol ingestion, metabolic derangement (AKA, DKA, lactic acidosis, upper GI bleed), and Boerhaave Syndrome. 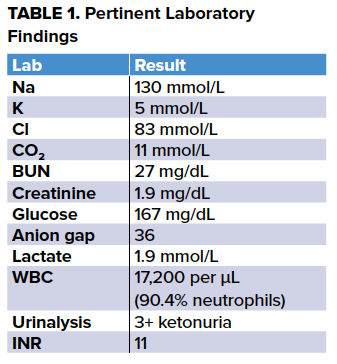
The remainder of the patient's laboratory evaluation - including liver enzymes, amylase, and lipase - were within normal limits, and methanol, ethylene glycol, salicylate, and digoxin levels were negative. Of note in the table above, the patient’s INR was greater than 11, above the upper limit of the assay, and this was confirmed by repeating the test. The patient showed no signs of bleeding.
The presumptive diagnosis of AKA was made, and the patient was treated with two, 250 ml boluses of 5% dextrose in normal saline spaced out over 1.5 hours given his history of heart failure, and then 5% dextrose in half normal saline at 75 mL/hour, 100 mg intravenous thiamine, and 10 mg of oral vitamin K. He was also placed on CIWA protocol while in the ED and received 1 mg of oral lorazepam. He was admitted to the internal medicine service for continued management. On hospital day one, after continued fluid resuscitation with 5% dextrose in half-normal saline, the patient’s anion gap closed, his INR decreased to 5.9, and he did not require lorazepam for treatment of alcohol withdrawal. By hospital day two, the patient’s INR normalized to therapeutic range and his warfarin was restarted. On hospital day three, the patient was discharged home with outpatient services for his alcohol use disorder.
Discussion
This case highlights the importance of diagnosing patients with AKA and providing the appropriate treatment. With early diagnosis and appropriate treatment, patients improve rapidly and serious complications are prevented.
Pathophysiology: Alcoholic ketoacidosis occurs through 3 metabolic pathways:
- Ethanol metabolism depletes NAD resulting in an elevated NADH/NAD ratio
- Volume depletion results in increased catecholamines and cortisol levels
- Starvation results in decreased glycogen stores
These three physiologic derangements create a state of catabolism with decreased insulin levels and increased glucagon levels resulting in inhibition of aerobic metabolism, ketone production (primarily β-hydroxybutyrate), and subsequent lipolysis.1,3
Diagnosis: Patients typically develop AKA after cessation of a prolonged episode of alcohol use. Clinically, patients experience nausea, vomiting, decreased PO intake, and abdominal pain but maintain a normal mental status. Lab values demonstrate a wide anion gap metabolic acidosis, with positive serum and/or urine ketones. Patient’s blood glucose levels can be low, normal, or mildly elevated.2 In AKA, high NADH/NAD causes pyruvate to be metabolized to lactate and volume depletion causes peripheral tissue hypoperfusion, resulting in a mild lactic acidosis.3 In the setting of altered mental status or markedly elevated blood glucose levels, clinicians should consider DKA rather than AKA as the diagnosis. In both AKA and DKA, the β-hydroxybutyrate level is expected to be in elevated; however, it is expected to be more significantly elevated in AKA in comparison with DKA.3
Treatment: Treatment for AKA requires glucose administration, thiamine supplementation, and volume repletion. Patients should be treated with 5% dextrose in normal saline until rehydrated, and then given 5% dextrose in half normal saline for maintenance. Importantly, volume repletion alone does not correct AKA as quickly as co-administration with dextrose, as dextrose administration stimulates insulin production, which halts lipolysis, and stops ketone production.4,5,6 One-hundred milligrams of thiamine should be given intravenously prior to dextrose administration to reduce the risk of Wernicke’s Encephalopathy and also to prevent further shunting of pyruvate to lactate. If the patient’s anion gap does not close with dextrose containing fluid administration, consider other causes of the patient’s anion gap including toxic alcohol ingestion. If patients are severely acidotic with a pH <7.0, clinicians can consider administering sodium bicarbonate. Patients with AKA often have electrolyte abnormalities which should be corrected accordingly. Cardiac arrhythmias associated with markedly abnormal electrolytes, including hypokalemia and hypomagnesemia, are a serious complication seen in patients with AKA.6 In patients with severe hypokalemia, it is important to replete potassium before administering any dextrose containing fluids, as this stimulates insulin production, causing an intracellular shift of potassium thus worsening their hypokalemia. It is important to recognize the difference between AKA and DKA, because patients with AKA treated with insulin develop severe hypoglycemia.
Why was this patient’s INR so high?
There was initial concern for acute liver failure until the patient’s hepatic function panel returned and argued against this diagnosis. Warfarin overdose was also considered, although the patient repeatedly denied this and reports he did not have access to his medications. Vitamin K deficiency was also considered. Vitamin K absorption is highly dependent on bile reabsorption, so patients with cholestatic liver disease, cirrhosis, or small bowel malabsorption are at risk for vitamin K deficiency.7,8 Additionally, adults with prolonged fasting or excessive vomiting can have vitamin K deficiency.9 Vitamin K deficiency can cause an elevation of INR due to poor function of vitamin K dependent coagulation factors, but will not affect PTT, platelet count, or fibrinogen level.9 One study found that patients with chronic alcohol use had abnormal carboxylation of prothrombin, resulting in abnormal prothrombin function. Subsequent vitamin K supplementation decreased levels of abnormal prothrombin in those same patients.10 Taken together, our patient’s INR may be elevated due to a vitamin K deficiency as a result of chronic alcohol intake and poor nutritional status, causing altered prothrombin function, resulting in a supratherapeutic INR. Further, vitamin K administration in our patient resulted in normalization of his INR.
Conclusion
Signs and symptoms of AKA can often be non-specific and should be considered in patients with recent cessation of heavy alcohol use with vomiting and metabolic derangements. It can be treated promptly with fluids, dextrose, and thiamine. An elevated INR in a patient with chronic alcoholism may be due to vitamin K deficiency, which has not been previously reported.
References
1. Wrenn KD, Slovis CM, Minion GE, and Rutkowski R. The Syndrome of Alcoholic Ketoacidosis. Am J Med. 1991;91:119-128.
2. Woods WA, Perina DG. Alcoholic Ketoacidosis. In: Tintinalli JE, Stapczynski J, Ma O, Yealy DM, Meckler GD, Cline DM. eds. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide. 8th ed. New York, NY: McGraw-Hill; 2016.
3. Yip L. Ethanol. In: Hoffman RS, Howland M, Lewin NA, Nelson LS, Goldfrank LR. Goldfrank's Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill; 2015.
4. Miller PD, Heinig RE, Waterhouse C. Treatment of alcoholic acidosis- the role of dextrose and phosphorous. Arch Intern Med. 1978;138:67-72.
5. Noor NM, Basavaraju K, Sharpstone D. Alcoholic ketoacidosis: A case report and review of the literature. Oxford Med Case Rep. 2016;2016(3):31-33.
6. McGuire LC, Cruickshank AM, Munro PT. Alcoholic ketoacidosis. Emerg Med J. 2006;23:417-420.
7. Lipsky JJ. Vitamin K deficiency. J Intensive Care Medicine. 1922;7:328-336.
8. Vitamin K. National Inistutites of Health. https://livertox.nih.gov/vitaminK.htm. Published May 9, 2016.
9. Absire TC. Abrams CS. Bleeding Risks and Vitamin K Deficiency. Transfusion Medicine and Hemostasis: Clinical and Laboratory Aspects (2e). 2013: 747-751.
10. Iberl FL, Shamszad M, Miller PA, Jacob R. Vitamin K deficiency in chronic alcoholic males. Alcoholism, clinical and experimental research. 1986;10(6):679-681.