



Torsades de pointes is a subcategory of polymorphic ventricular tachycardia, and case reports have linked torsades de pointes with hemochromatosis due to elevated iron stores.
Case
A 33-year-old female with past medical history of hypothyroidism, questionable hemochromatosis, and alcohol use disorder presented to the emergency department (ED) with a chief complaint of palpitations. She was at an appointment with her hematologist when she began to feel lightheaded and experienced palpitations. Vital signs at the clinic revealed tachycardia with a rate >200 BPM along with borderline hypotension. Therefore, she was sent to the ED for further evaluation.
Prior surgeries include herniorrhaphy. She reported using 1 pack of cigarettes per day, a history of alcohol use, and no recreational drug use. Her prescription medications included levothyroxine, hydroxyzine, and omeprazole.
On arrival to the ED, the patient had laboratory results from her hematologist from several days prior, which were notable for a potassium of 2.8 mEq/L. She stated that she had previous issues with hypokalemia of unclear etiology. Initial vital signs in the ED included HR 99 BPM, BP 98/53, otherwise normal. EKG in triage showed a rate of 142 BPM and multiple runs of wide complex tachycardia consistent with ventricular tachycardia with intermittent supraventricular beats (Figure 1). She was immediately brought to a room and placed on the cardiac monitor. She continued to have 5-10 second runs of monomorphic ventricular tachycardia followed by several seconds of normal sinus rhythm. She then developed nausea and was given 4 mg IV ondansetron.
Figure 1. Initial EKG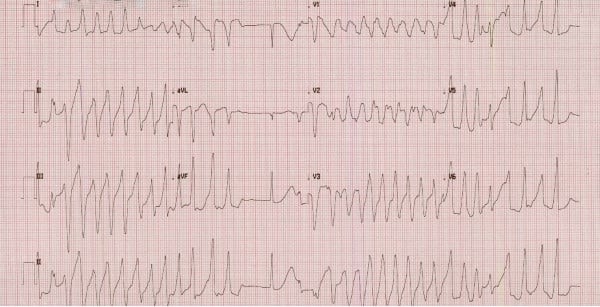
While labs were being drawn, defibrillator pads were applied, and she was given 10 mEq IV potassium chloride given her known hypokalemia. She was given a bolus of 150 mg amiodarone followed by an amiodarone drip of 1 mg/minute. She also received 2g magnesium sulfate given the known relationship between hypokalemia and hypomagnesemia. After several minutes, repeat EKG revealed normal sinus rhythm with a long QT interval of >500 msec (Figure 2).
Figure 2. Repeat EKG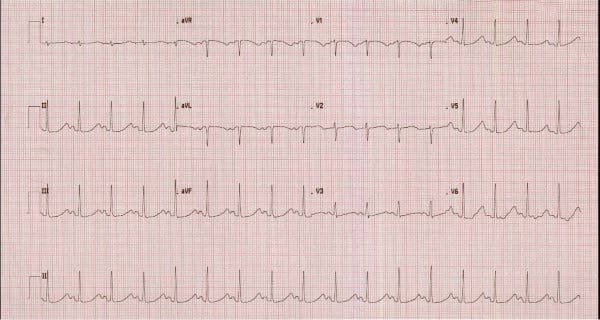
In further discussion with the patient, she revealed that she did have a history of a long QT interval in the past which was attributed to hypothyroidism, but that her QT interval reportedly normalized at that time. Her labs in our ED were significant for a potassium of 2.7 mEq/L, mildly elevated total bilirubin of 1.7 and AST of 85 with normal ALT of 40. Her CBC was unremarkable. Her troponin was not elevated. Her TSH was elevated at 3.95 with a normal free T4 level of 0.90. After her labs resulted, her potassium was repleted further with 40 mEq IV KCl and 30 mEq PO KCl. Cardiology was consulted and agreed with continuing the amiodarone drip and aggressively repleting her potassium.
Several hours later while waiting for an inpatient bed, she became unresponsive, cyanotic, and pulseless. Cardiac monitor revealed polymorphic ventricular tachycardia consistent with torsades de pointes. Chest compressions were performed, and the patient was defibrillated once at 200 J biphasic. She regained pulses and returned to her normal level of consciousness after a few minutes. She did not require intubation. Cardiology was again called, who advised discontinuing the amiodarone drip and recommended giving her a lidocaine bolus followed by a continuous lidocaine infusion; 2g IV magnesium sulfate was also administered. She was then admitted to the ICU for further workup and close monitoring.
Discussion
Torsades de pointes is a subcategory of polymorphic ventricular tachycardia. It is associated with long QT syndrome and characterized by the unique appearance of small and large amplitude of complexes that appear as though a ribbon is being “twisted around a point.” Common causes include electrolyte abnormalities (hypokalemia, hypomagnesemia, hyperphosphatemia, hypocalcemia), Class 1A antiarrhythmics (procainamide, quinidine, disopyramide), and other medications (many psychotropic medications, antibiotics, antiemetics). Some patients with long QT syndrome have a congenital cardiac ion channel defect that results in a long QT interval.
Interestingly, there have been cases of long QT and torsades de pointes associated with hemochromatosis due to elevated iron stores. It has been hypothesized that even in patients without any known genetic cause of long QT syndrome, without any electrolyte derangements, and without any structural cardiac abnormalities, significantly elevated iron stores may result in iron deposition in cardiac tissue. Cardiac hemochromatosis may result in a long QT interval and progress to polymorphic ventricular tachycardia due intramuscular conduction delay.3 Animal models have also correlated elevated iron stores with increased rates of sudden cardiac death. Ferric iron has been shown to disturb calcium and potassium channels, particularly in phase 2 of the cardiac action potential, which corresponds directly to the length of the QT interval.4
Case Conclusion
While in the hospital, the patient did not have any additional ventricular tachydysrhythmias. She was treated with beta-blockers and about 36 hours of IV lidocaine infusion. An inpatient echocardiogram was unremarkable, and cardiology had low suspicion for contributing cardiac ischemic events. She was fitted for a LifeVest prior to discharge and was referred for more comprehensive genetic testing regarding congenital long QT syndrome and possible need for an implanted defibrillator.
Review of the patient’s records show that about 5 weeks prior to her presentation in the ED with ventricular tachycardia, her ferritin level was severely elevated >1650 ng/mL (normal range 10-291). She had not yet had any phlebotomy or pharmacologic treatment for iron overload between that lab measurement and her cardiac dysrhythmia.
She was also encouraged to reschedule her phlebotomy appointment with her hematologist given the possible contribution of iron overload on dysrhythmias. While she did test negative for three different genotypes that have been associated with hemochromatosis (H63D, C282Y, and S65C), there may be genotypes not yet identified that would correlate with hemochromatosis and explain her iron overload.
This patient did also undergo the aforementioned genetic testing required to assess whether she has congenital long QT syndrome. Her workup showed she had an autosomal dominant mutation in a gene KCNEI D76N, which is associated with potassium channel defects in cardiac tissue that can lead to a prolonged QT interval.6
This patient had multiple factors that likely contributed to her development of ventricular tachycardia and ultimately torsades de pointes. She presented with severe hypokalemia and hypomagnesemia in addition to having severely elevated iron stores and carrying a mutation in a gene that encodes cardiac potassium channels, all of which increased her risk of prolonged QT interval and progression to torsades de pointes.1-3 In addition, she had received amiodarone, which is generally a good choice for treatment of ventricular tachycardia and appeared to initially stabilize the patient. However, amiodarone may also increase QT interval, and thus should not be the first choice once established that a patient has a prolonged QT interval.5 In such scenarios, electrolyte correction, magnesium infusion, and lidocaine are the mainstays of treatment. In the case of patients with underlying hemochromatosis, emergent phlebotomy and deferoxamine treatment may also be considered.3
References
- Demant AW, Schmiedel A, Büttner R, Lewalter T, Reichel C. Heart failure and malignant ventricular tachyarrhythmias due to hereditary hemochromatosis with iron overload cardiomyopathy. Clin Res Cardiol. 2007;96(12):900-903.
- Fujino T, Inoue S, Katsuki S, Higo T, Ide T, Oda Y, Tsutsui H. Fatal cardiac hemochromatosis in a patient with hereditary spherocytosis. Int Heart J. 2018;59(2):427-430.
- Lee N, Park J, Rhee K. Acquired long qt syndrome and sudden cardiac death due to secondary hemochromatosis with multitransfusions for severe aplastic anemia. Ann Hematol. 2008;87(11):933-935.
- Refaat MM, El Hage L, Steffensen AB, Hotait M, Schmitt N, Scheinman M, Badhwar N. Iron overload leading to torsades de pointes in Β-Thalassemia and Long Qt Syndrome. Card Electrophysiol Clin. 2016;8(1):247-256.
- Schofer JM, Sunga KL, Colletti JE. Emergency medicine: a focused review of the core curriculum. Cardiovascular Disorders. In: Emergency Medicine: a Focused Review of the Core Curriculum. 2nd ed. Milwaukee, WI: American Academy of Emergency Medicine Resident and Student Association; 2018:71-74.
- Splawski I, Shen J, Timothy KW, et al. Spectrum of mutations in long-qt syndrome genes. Circulation. 2000;102(10):1178-1185.